11.МЕТОДИ ВИЗНАЧЕННЯ ФОСФОРОВМІСНИХ СПОЛУК І ІНШИХ ЛІПІДІВ
11.1 Спектрофотометричне визначення нуклеїнових кислот
Рослина
є досить складним
об'єктом для дослідження нуклеїнових кислот, проте в даний час є чимало методів
їх кількісного визначення в різних тканинах, які дозволяють визначати РНК і ДНК
в одній
наважці.
Визначення фосфору РНК і ДНК ведеться на спектрофотометрі за
довжини
хвиль 270 і 290 нм в свіжому або фіксованому матеріалі.
Фіксацію матеріалу можна проводити ліофілізацією або гарячим 96 % - вим етанолом. Попереднє видалення пігментів, кислоторозчинного і ліпідного фосфору є неодмінною умовою спектрофотометричного визначення фосфору РНК і ДНК.
Суть методу
Полягає у екстракції нуклеїнових кислот із біологічного матеріалу
гарячою перхлоридною кислотою з подальшим визначенням поглинання екстрактів в
ультрафіолетовій області спектра при 270 і 290 нм. Інтенсивність
світлопоглинання пропорційна кількості нуклеїнових кислот у пробах матеріалу.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали:
терези аналітичні, роторний випарювач, центрифуга,
морозильна камера, холодильна камера, ексикатор (або вакуум-ексикатор),
скляний фільтр №4 (або знезолений "дрібнопористий" фільтр), колби
конічні, мірні циліндри, мірні колби на 50, 100 см3, піпетки,
бюретки, широкі пробірки, порцелянова ступка з товкачиком, термостат (або
термокамера з забезпеченням постійної температури 37°С), водяна та льодяна баня, повітряний зворотний холодильник.
Реактиви: 0,5 н перхлоридна кислота HClO4, 57 % - ва
перхлоридна кислота HClO4, 5 % - ва перхлоридна
кислота HClO4, 0,2 н NaOH, 0,5 н KOH (або NaOH), кріогідратна
суміш, вода дистильована, фенолфталеїн, суміш етанолу і етилсульфату (3:1),
96% - вий етанол, скляний пісок.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Хід аналізу:
- 5…7 г досліджуваного матеріалу поміщають в широку
пробірку;
- заливають 10…15 см3 0,5 н перхлоридною кислотою
HClO4, яка охолоджена до 0 ° С;
- пробірку з наважкою поміщають в кріогідратну суміш (сухий лід, холодний метанол або рідкий азот) для фіксації;
- після фіксації наважку з HClO4 переносять в порцелянову
ступу, ретельно розтирають зі скляним піском до гомогенного стану і прокачують
на роторі 30 хв. при низькій температурі;
- отриманий гомогенат центрифугують протягом 5 хв. при 3000
об / хв.;
- екстракт зливають і ставлять в холодильник;
- осад заливають 10 см3 0,5 н HClO4 і прокачують ще
30 хв. при 0 ° С,
- потім центрифугують за тих же умов;
- екстракти об'єднують в мірній колбі або циліндрі, а осад тричі
промивають холодною дистильованою водою;
- промивні води об'єднують з кислотними екстрактами;
- негайно нейтралізують 0,2 н NaOH по фенолфталеїну і доводять до певного
об'єму;
- утворені пластівці натрієвої солі хлоридної кислоти видаляють
центрифугуванням;
- нейтралізований хлорнокислий екстракт може бути використаний для
визначення вільних аденілових нуклеотидів;
- осад після видалення кислоторозчинного фосфору тричі обробляють
96% - вим етанолом на холоді;
- потім проводять багаторазову екстракцію сумішшю етанол-етилсульфату (3:
1) за кімнатної температури до повного вилучення пігментів;
- останній раз осад промивають етилсульфатом і висушують в ексикаторі
(краще вакуум-ексикаторі);
- підготовлена таким чином рослинна маса використовується для визначення
нуклеїнових кислот;
- осад після вилучення кислоторозчинного і ліпідного фосфору заливають
0,5 н KOH (або NaOH) з розрахунку 10 см3 лугу на 100 мг осаду і
витримують при 37 ° С протягом години;
- потім охолоджують і центрифугують 15 хвилин при 4000
об/хв.;
- надосадову рідину зливають і
ставлять в холодильник, а осад після промивання холодним 0,5 н КОН
відкидають;
- прозорі центрифугати об'єднують і нейтралізують на крижаній бані
холодною 57 % -вою перхлоридною кислотою HClO4 за
фенолфталеїном;
- потім концентрацію розчину цієї кислоти доводять до 5% її
вмісту;
- на холоді випадає осад ДНК, а в розчині залишається
РНК;
- через 20 хв. осад відокремлюють центрифугуванням і два рази (по 5
см3) промивають охолодженою 5 % - вою
HClO4;
- центрифугати, що містять РНК, об'єднують в конічної колбі на 50
см3;
- закривають зворотним повітряним холодильником і гідролізуют на киплячій
водяній бані протягом 15 хв.;
- потім охолоджують і фільтрують через скляний фільтр №4 або через
знезолений "дрібнопористий" фільтр;
- об'єм прозорого фільтрату вимірюють і використовують для
спектрофотометричного визначення фосфору РНК;
- осад ДНК за допомогою 5 % - вої HClO4 переносять в конічну колбу
на 50 см3;
- закривають зворотним повітряним холодильником і гідролізуют 30 хв. на
киплячій водяній бані;
- після охолодження і центрифугування рідину зливають в мірну колбу, а
осад промивають невеликою кількістю 5 % - вої HClO4, центрифугують і надосадову рідину приєднують до
першого центрифугату;
- об'єм доводять до мітки або просто вимірюють;
- прозорий розчин спектрофотометрують: в якості контролю, проти якого
вимірюють оптичну густину дослідного розчину, беруть
5 % - вий розчин HClO4;
- оптичну густину отриманих дослідних розчинів вимірюють при 270 і 290 нм.
Розрахунки:
Обчислення концентрації фосфору нуклеїнових кислот (Х) проводять за
формулою:

де
Е – оптична густина розчинів при відповідній довжині хвилі.
Ділення на 190 дає кількість фосфору нуклеїнової кислоти в 1 см3 розчину.
Приклад
розрахунку:
При
спектрофотометричному аналізі
розчину РНК знайдена Е270 = 836, а Е290 –
506. Об'єм
розчину РНК становив 100 см3.
Перед визначенням оптичної густини
він був розбавлений в 3 рази. Наважка
рослинного матеріалу в перерахунку на суху вагу 0,5 г. Кількість
фосфору РНК буде:
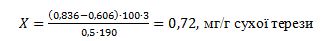
11.2 Визначення вмісту неорганічних поліфосфатів (за Р. Лангену, Р. Ліссу, модифікація І. С. Кулаева та ін.)
В обміні речовин багатьох нижчих організмів істотне місце займають перетворення неорганічних поліфосфатів, які є макроергічними сполуками. Серед методів, що використовують для кількісного визначення цих речовин, у даний час найбільш вдалим є метод Ланге і Лісса, модифікований І. С. Кулаєвим з співробітниками.
Суть методу
Полягає у
визначенні
кількох фракцій неорганічних поліфосфатів, які
розрізняються молекулярною вагою, внутрішньоклітинною
локалізацією,
а також фізіологічним значенням.
Даний метод дозволяє враховувати п'ять різних фракцій поліфосфатів: кислоторозчинні, солерозчинні, лужнорозчинні при рН 8…9, лужнорозчинні при рН 12 і поліфосфати, які екстрагуються HClO4 при 90…100°С. Крім того, ця методика дає можливість оцінити характер накопичення фосфору в деяких органічних сполуках. Так, органічний фосфор кислоторозчинної (після сорбції нуклеотидів) і солерозчинної фракцій, представлений, головним чином, фосфором вуглеводів. Отже, органічний фосфор обох фракцій може відображати характер накопичення фосфовуглеводів. Фосфор ліпідів можна виявити за фосфором фракцій, які витягуються сумішшю етанол-етеру. Органічний фосфор лужнорозчинних фракцій і фракції гарячого кислотного екстракту містить до 95% фосфору нуклеїнових кислот. Тому органічний фосфор цих трьох фракцій дозволяє судити про характер накопичення нуклеїнових кислот. Залишок за даних умов фракціювання містить головним чином фосфопротеїни, отже, величина залишку фосфору може характеризувати накопичення фосфопротеїнів.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали:
терези аналітичні, роторний випарювач, центрифуга,
морозильна камера, холодильна камера, ексикатор (або вакуум-ексикатор),
скляний фільтр №4 (або знезолений "дрібнопористий" фільтр), колби
конічні, мірні циліндри, мірні колби на 50, 100 см3, піпетки,
бюретки, широкі пробірки, порцелянова ступка з товкачиком, термостат (або
термокамера з забезпеченням постійної температури 37°С), водяна та льодяна баня, повітряний зворотний холодильник.
Реактиви: 0,5 н перхлоридна кислота HClO4, 57 % -ва
перхлоридна кислота HClO4, 5 % - ва перхлоридна
кислота HClO4, 0,2 н NaOH, 0,5 н KOH (або NaOH), кріогідратна
суміш, вода дистильована, фенолфталеїн, суміш етанол-етилсульфату (3:1),
96% - вий етанол, скляний пісок.
ацетатний буфер: 9 г натрій ацетату та 0,5 см3 купрум
(ІІ) сульфату доводять до 100 см3 4 и оцтовою
кислотою;
розчин молибдоату: 5 % - вий розчин амоній молібдату у воді;
метольний реактив: 0,2 % - вий розчин метолу у
5 % - вому розчині натрій сульфату.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Хід аналізу:
1)Кислоторозчинна
фракція:
- наважку
досліджуваного матеріалу (5…7
г) заливають 10…15
см3
0,5 н НСlО4 і фіксують у
кріогідратній
суміші;
- після
фіксації наважку
з НСlО4 переносять у
порцелянову ступку, розтирають з кварцовим піском до гомогенного
стану;
- прокачують на ротарі 30 хв.
при низькій температурі;
- отриманий
гомогенат центрифугують протягом 5 хв.
при 3000 об/хв;
- екстракт
зливають і ставлять в холодильник;
- осад
заливають 10 см3
0,5 н НСlО4;
- прокачують ще раз протягом 30 хв.
при 0°С;
- центрифугують
за
тих же умов;
- екстракти
об'єднують у
мірній колбі, а осад тричі промивають холодною дистильованою водою;
- промивні
води об'єднують з кислотними екстрактами;
- негайно
нейтралізують 0,2 н NaOH по фенолфталеїну і доводять до певного об'єму;
- утворені пластівці натрієвої солі хлоридної кислоти видаляють центрифугуванням;
Для
кількісного визначення поліфосфатів проводять такі операції:
- визначають оптичну густину екстракту при 260 нм;
- проводіть сорбцію нуклеотидів на активоване вугілля марки БАУ з розміром
частинок 0,5…1,0 мм або марки Norit
A (Голландія). Сорбцію проводять на холоді при постійному перемішуванні
протягом 1…2 годин.
- потім
вугілля відокремлюють або центрифугуванням, або фільтруванням;
- в екстракті знову визначають оптичну густину при 260 нм і, якщо вона
становить понад 10% від початкової, обробку вугіллям повторюють;
- після видалення нуклеотидів в екстракті визначають: 1) ортофосфат (Рорто) безпосередньо в екстракті; 2) фосфор сполук, які гідролізуються до ортофосфата протягом 7 хвилин (Р7) і 30 хвилин (Р30); 3) загальний фосфор (Рзаг) після спалювання частини екстракту з концентрованою Н2SO4 і застосуванням в якості каталізатора НСlО4. Кількість поліфосфатів розраховують за формулою Ломана і Ланге:
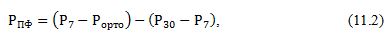
де
величина (Р7
–
Рорто)
являє собою лабільний фосфор фракції.
Вміст
стабільного фосфору, який представлений фосфором органічних сполук, знаходять
за
різницею
між загальним фосфором і сумою фосфору поліфосфатів та
ортофосфата:
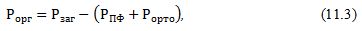
2)Солерозчинна
фракція:
- після
видалення кислоторозчинних з'єднань до залишку додають 1,5 г натрій перхлорату
і 1 см3
0,5 н НСlО4;
- прокачують
на холоді протягом 15 хв.
- після
цього додають 15 см3
холодної дистильованої води і продовжують екстракцію ще 15 хв.;
- екстракт
відокремлюють центрифугуванням при 3000 об/хв.,
- дана процедура повторюється
двічі;
- осад
промивається кілька разів холодною дистильованою водою;
- промивні
води об'єднують з екстрактами і доводять до певного об'єму;
- солерозчинну
фракцію аналізують так само, як і кислоторозчинну.
3)Фракція
фосфоліпідів:
- Залишок
після вилучення солерозчинної
фракції
тричі обробляють 96 % - вим
етанолом на холоді;
- потім
проводять багаторазову екстракцію сумішшю етанол-етилсульфату (3:1)
за
кімнатної
температури до
повного вилучення пігментів;
- останній
раз залишок промивають етером і висушують в вакуум-ексикаторі;
- всі спиртові і спирт-етерні екстракти об'єднують в мірній колбі і
доводять до мітки;
- у цій фракції визначають загальний фосфор після спалювання частини
екстракту в концентрованій H2SO4 і НСlО4;
- за загальним фосфором фракції судять про вміст фосфоліпідів:

4)Лужнорозчинна фракція, рН 8…9.
- до залишку після видалення ліпідів додають кілька см3
холодної дистильованої води;
- за допомогою 0,2
н NaOH доводять рН розчину до 8…9 при 0°С;
- екстракцію
проводять протягом 40 хв.
при постійному перемішуванні;
- потім
екстракт відокремлюють центрифугуванням;
- до
залишку додають невелику кількість холодної дистильованої води і залишають ще на
40 хв.;
- екстракт
знову відокремлюють центрифугуванням;
- осад
ретельно промивають холодною водою;
- обидва
екстракти
об'єднують з промивними водами і доводять до певного об'єму;
- у фракції визначають: 1) ортофосфат (Рорто); 2) фосфор сполук, які гідролізуються до ортофосфата протягом 7 хвилин (Р7); 3)Загальний фосфор (Рзаг).
- кількість поліфосфатів розраховують за величиною лабільного фосфору фракції:
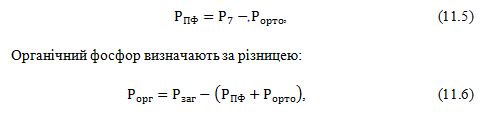
5)Лужнорозчинна фракція, рН 12:
- цю фракцію екстрагують 0,05 н NaOН (pH = 12) при 0 ° С і
ретельному перемішуванні протягом 2 годин;
- екстракт відокремлюють центрифугуванням, а залишок кілька разів
промивають холодною дистильованої водою;
- промивні води об'єднують з екстрактом і доводять до певного
об'єму;
- фракцію аналізують так само, як і попередню.
6)Фракція,
яка
екстрагуються НСlО4 при 90-100°С:
- залишок після видалення лужнорозчинних фракцій обробляють 0,5 н
НСlО4 на водяній бані при 90…100°С послідовно протягом 20 і 10 хв.;
- залишок
після центрифугування ретельно промивають холодною дистильованою
водою;
- екстракти
об'єднують з промивними водами і доводять до певного об'єму;
- в
об'єднаному екстракті
визначають: 1) ортофосфат; 2) загальний фосфор;
- вважається, що ортофосфат цієї фракції утворюється в основному в
результаті гідролізу неорганічних поліфосфатів. Тому
про кількість поліфосфатів у даній фракції судять за величиною ортофосфата:
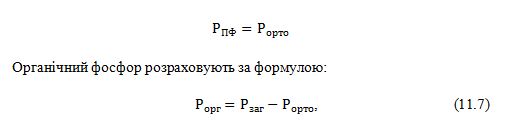
7)Фракція
фосфопротеїнів:
- після всіх екстракцій залишок спалюють з концентрованою H2SO4 і HClO4 і визначають загальний фосфор;
за
загальним фосфором
судять про вміст
фосфопротеїнів:
При кількісному обліку неорганічних поліфосфатів визначення фосфору можна проводити за методом Пануша. При використанні цього методу може відбуватися частковий гідроліз кислотолабільних з'єднань, але проте він простий і швидкий.
Хід аналізу:
- до 2,75 см3 розчину, який містить від 20 до 200 нМ фосфату,
додають 1,25 см3 ацетатного буфера pH (9 г натрій ацетату і 0,5 см3 купрум (ІІ) сульфату доводять
до 100 см3 4 н оцтовою кислотою), 0,5 см3 розчину
молибдата (5 % - вий розчин амоній молібдату у воді) і 0,5 мл метольного реактиву
(0,2 % - вий розчин метола у 5 % - вому розчині натрій
сульфату);
- через
60 хв.
вимірюють поглинання розчину на спектрофотометрі при довжині хвилі 960 нм з
киснево-кадмієвим
елементом;
- кількість
фосфору в пробі визначають за
калібрувальною
кривою;
- для визначення Р7 і Р30 до 1 см3 проби
додають 1 мл 2 н HCl, розчин гідролізують 7 і 30 хв. відповідно при 100°С, після чого швидко охолоджують і проводять визначення фосфору, як
описано вище;
- для визначення загального фосфору до частини проби додається 0,5 см3 концентрованої HClO4 і 10 см3 H2SO4; після упарювання при 120°С проба спалюється при 200°С до повного знебарвлення; нейтралізовану після спалювання пробу доводять до певного об'єму і визначають ортофосфат.
11.3 Визначення вмісту
жирів у свіжій та консервованій плодоовочевій
продукції
Вміст жирів у фруктах та овочах є незначним. Значно більше їх
накопичується в насінні. В ядрах зерняткових та кісточкових плодів і баштанних
овочів кількість жирів становить в середньому 20…60 %. Жири можуть
накопичуватися в листках, стеблах, коренях, частинах квітки, але вміст їх не
перевищує 5 %.
Чисті рослинні жири – олії – безбарвні речовини. Вони нерозчинні у воді
та у клітинах знаходяться у формі диспергованих в протоплазмі крапель. До складу
рослинних жирів може входити до 50 різних жирних кислот.
Вміст жиру нормується і контролюється також у напівфабрикатах і готовій
продукції. У консервах він повинен бути не менше: овочі обезжирені в томатному
соусі – 6 %, баклажани – 8 %, порізані кружечками кабачки – 8 %, овочева
ікра – 9 %.
Жир виділяють екстрагуванням, центрифугуванням. Екстрагування засноване
на тому, що жири нерозчинні у воді, але добре розчиняються в органічних
речовинах, таких як хлороформ, бензин, етиловий і пертолейний етер.
Найчастіше як розчинник використовують етиловий етер: його відгіняють, а за залишком визначають вміст жиру в продукті. Щоб в екстракт не переходили інші речовини, етер попередньо очищують від домішок спирту, води, а жир визначають у просушеному продукті.
1.1.1
Визначення вмісту жиру у свіжій
сировині
Метод розповсюджується на свіжу
плодоовочеву сировину, горіхи та насіння з невисокою олійністю.
Суть методу
Полягає у швидкому (протягом 4…5 хв.) добуванні жиру із продукту бензином в металевому екстракторі з
подальшим визначенням маси жиру в аліквотній частини отриманого екстракту після
видалення розчинника.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали:
аналітичні терези, екстрактор-подрібнювач, термометр, алюмінієва чашка
об'ємом 8…10 см3, піщана баня, ексикатор, градуйована піпетка,
фільтрувальний папір, конічні та мірні колби.
Реактиви: безводна кальцинована сода, або NaO, або NaHPO4, бензин.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до
аналізу виконуюють згідно пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Хід аналізу:
- у попередньо зважений пакет із фільтрувального паперу (4х10 см),
виготовлений у вигляді кишені зважують на терезах 10 г підготовленої середньої
проби матеріалу;
- наважку переносять на дно металевої пробірки
екстрактора-подрібнювача;
- додають 7…8 г безводної
кальцинованої соди (або NaO,
або NaHPO4);
- додають 20…25 см3 бензину (з температурою кипіння 120
°С);
- суміш ретельно розтирають металевим товкачиком протягом 4…5
хв.;
- сода, поглинаючи воду, виділяє тепло, яке сприяє екстрагуванню жиру;
необхідно стежити, щоб температура не піднімалась вище 32 °С, оскільки сода
втрачає здатність поглинати вологу; пробірку за необхідності
охолоджують;
- після розмішування та розчинення розчин фільтрують: в металеву пробірку
вставляють пробку з трубкою для фільтрування, в якій попередньо встановлено
градуйовану піпетку; на кінці відвідної трубки закріплена резинова груша, під
дією якої повітрям розчин жиру подається в піпетку;
- залежно від концентрації жиру встановлюють об'єм фільтрату 2…5
см3;
- обережно, по краплям з піпетки переносять фільтрат у попередньо висушену
та зважену алюмінієву чашку;
- нагрівають на піщаній бані за температури
180…200°С;
- після видалення розчинника чашку з жиром охолоджують в ексикаторі
протягом 1…3 хв.,
- зважують з точністю до 0,01 г.
Розрахунки:
Вміст жиру Х, %, визначають за формулою:
де m – маса наважки, г,
А –
кількість розчинника, взятого для добування жиру, см3,
Б – маса
жиру в чашці, г,
0,92 – коефіцієнт
рослинної олії чи свінячого жиру, для інших жирів – 0,94.
За кінцевий результат приймають середнє арифметичне двох паралельних
повторень. Різниця між паралельними визначеннями не повинна перевищувати 0,5%.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
1.
2.
3.
i.
1.1.2
Визначення масової частки жиру в продуктах переробки
плодів і овочів гравіметричним та рефрактометричним методами
Методи поширюється на продукти переробки плодів і овочів, включаючи
продукти харчування з картоплі.
Суть методу
Полягає у екстракції жиру сумішшю хлороформу
і етилового спирту в фільтруючій ділильній лійці з подальшим визначенням його
маси в отриманому екстракті після видалення
розчинника.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: терези лабораторні загального призначення з найбільшою межею зважування
до 200 г не нижче 3-го класу точності; шафа сушильна електрична, годинники
піскові настільні на 1 і 2 хв.; піпетка на 5 см3; циліндри місткістю
10, 25 і 50 см3; установка для екстрагування жиру (рис.34), баня
водяна, насос водоструминний, ексикатор з осушувачем - прожареним хлористим
кальцієм або сульфатною кислотою, густиною 1,84 кг/м3 ,
стаканчики для зважування (бюкси), стакан місткістю 10 см3; палички з
хіміко-лабораторного скла, лійка лабораторна, лійка ділильна місткістю 1000
см3, щипці тигельні.
Реактиви: спирт етиловий ре
ктифікований технічний, хлороформ технічний, кислота нітратна, х.ч. густиною 1,41 г/см3; кислота сульфатна, х.ч. густиною 1,84 г/см3; кислота оцтова, х.ч. густиною 1,07 г/см3; натрій хлористий, ч.д.а.; вода дистильована.
Підготовка до проведення вимірювань
- відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1. Проби, які надходять у лабораторію повинні бути без пошкоджень і
змін якості продукту при транспортуванні та зберіганні.
- бюкси після миття сушать в сушильній шафі, охолоджують в ексикаторі і
зважують;
- готують суміш для екстракції, змішуючи два об'єми хлороформу з одним об'ємом етилового спирту;
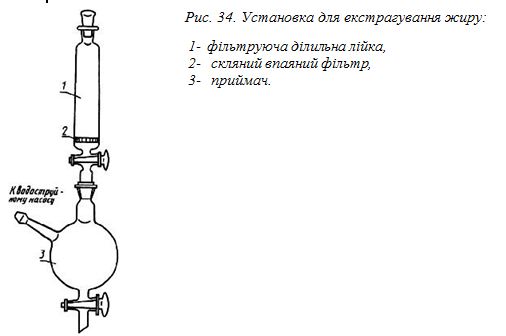
- збирають установку для екстрагування жиру відповідно до рисунку 34;
- після багаторазового використання фільтруючої ділильної лійки в разі
уповільнення швидкості фільтрації проводять регенерацію скляного фільтра:
промивають лійку водою, заливають 50…100 см3 суміші з рівних об'ємів
нітратної та сульфатної кислот і залишають з відкритим краном для стікання
суміші. Через 10…12 годин лійку промивають проточною водопровідною водою,
з'єднують з приймачем і знову промивають при включеному насосі послідовно 30
см3 дистильованої води, а потім 30 см3 екстрагуючої
суміші.
- у приймач вносять 2…3 см3 екстрагуючої суміші.
Хід аналізу:
- беруть наважку масою близько 2,0 г з гомогенізованої середньої проби
продукту;
- за допомогою 5 см3 етилового спирту скляною паличкою наважку
кількісно переносять у фільтруючу ділильну лійку;
- якщо продукт містить молочний жир, в лійку з наважкою додають 2 краплі
оцтової кислоти і суміш витримують 10 хв.;
- доливають 20 см3 екстрагуючої суміші;
- закривають лійку пробкою і струшують вміст протягом 2
хв.;
- приєднують лійку до приймача, включають водоструминний насос і
відсмоктують отриманий екстракт жиру;
- повторюють екстракцію ще двічі, доливаючи до наважки у лійці по 15
см3 екстрагуючої суміші і струшуючи лійку протягом 1
хв.;
- екстракти з приймача за допомогою 10 см3 екстрагуючої суміші
кількісно переносять в ділильну лійку місткістю 1000
см3;
- додають 100 см3 дистильованої води;
- вносять 3 г хлористого натру;
- струшують лійку з вмістом 2 хв.;
- після поділу шарів нижній хлороформенний шар зливають у
бюкс;
- бюкс з екстрактом поміщають на водяну баню і випарюють розчинник до
зникнення його запаху;
- потім поміщають бюкс з залишком в сушильну шафу, нагріту до (100
± 5) °С;
- сушать за цієї температури протягом 10 хв., охолоджують 25…30 хв. в ексикаторі і зважують.
Розрахунки:
Масову частку жиру Х, в %, обчислюють за формулою:

де m1- маса жиру, г;
m - маса наважки продукту, г.
За остаточний результат випробування приймають середнє арифметичне результатів двох паралельних визначень, допустима абсолютна розбіжність між якими не повинна перевищувати 0,1% при масовій частці жиру до 2%; 0,3% - при масовій частці жиру від 2 до 5%; 0,5% - при масовій частці жиру від 5 до 8%; 0,7% - при масовій частці жиру від 8 до 15%; 1,1% - при масовій частці жиру від 15 до 30% і 1,5% - при масовій частці жиру понад 30%.
11.3.2.2 Рефрактометричний метод визначення жиру
Суть методу
Полягає в екстракції жиру 1-бромнафталіном з
подальшим визначенням показника заломлення
екстракту.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: подрібнювачі тканин, ексикатор, терези, скло годинне, палички з
хіміко-лабораторного скла, папір фільтрувальний, вата, натрій сульфат,
скальпель, рефрактометр, шкала якого градуйована в одиницях показника
заломлення, з межами вимірювання 1,3…1,7, ціною поділки не більше 0,001 і
границями основної допустимої похибки ± 0,0005; засоби для забезпечення
циркуляції води і підтримки температури призм рефрактометра постійної в межах ±
0,2 °С близько 20 °С; термометр рідинний скляний з межею допустимої похибки не
більше ± 0,3 °С в діапазоні температур 10…30 °С, пробірка місткістю 5 або 10
см3; годинники піскові настільні на 3 хв.; ступка і
товкачик.
Реактиви: спирт етиловий, 1-бромнафталін, ч.; етер етиловий технічний; пісок
кварцовий.
Підготовка до проведення вимірювань
- відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1. Проби, які надходять у лабораторію повинні бути без пошкоджень і
змін якості продукту при транспортуванні та зберіганні;
- протирають призми рефрактометра ватою, змоченою водою, спиртом або
етером, сушать і перевіряють установку нуль-пункту за дистильованою водою
відповідно до інструкції по експлуатації приладу;
- визначають показники заломлення 1-бромнафталіну і жиру при 20 °С
(n20)
- вимірювання допускається проводити при температурі, відмінній від 20 °С,
в інтервалі 15…25 °С, але при цьому слід вносити поправку на температуру
вимірювання, яку фіксують з похибкою не більше 0,2 °С;
- показник заломлення за температури вимірювання, відмінною від 20 °С, обчислюють за формулою:

де n20- показник заломлення за температури вимірювання;
Δt - поправочний коефіцієнт (Δt = 0,000440 ° С-1 для 1-бромнафталіну,
Δt = 0,000346 ° С-1 для
жиру);
t - температура вимірювання, ° С.
Показник заломлення 1-бромнафталіну (n20= 1,658) перевіряють для кожної нової партії реактиву, а також
періодично при тривалому зберіганні. За необхідності реактив зневоднюють,
витримуючи його в ексикаторі з осушувачем.
Показник заломлення жиру перевіряють для кожного продукту. Для цього близько 10 г продукту змішують з 20 см3 етилового етеру, екстракт фільтрують, з фільтрату відганяють етер і отриманий жир використовують для рефрактометрії.
Хід аналізу:
- беруть наважку масою близько 5,0 г із гомогенізованої проби
продукту;
- кількісно переносять в порцелянову ступку скальпелем за допомогою 4 г
натрій сульфату;
- додають 2 г піску і вносять від 5,5 до 6,5 г (близько 4 см3)
1-бромнафталіну, встановлюючи точне значення його маси по різниці між масою
пробірки з реактивом і масою пробірки із залишками
реактиву;
- суміш розтирають товкачем близько 3 хв.;
- фільтрують через паперовий фільтр;
- перші 2…3 краплі фільтрату відкидають, а наступні 2 краплі наносять на
призму рефрактометра;
- визначають показник заломлення екстракту при
20 ° С;
- при проведенні вимірювань в інтервалі температур 15…25 ° С показник заломлення обчислюють з урахуванням температури вимірювання за формулою 11.10, приймаючи Δt = 0,000434 ° С-1.
Розрахунки:
Масову частку жиру Х, %,
обчислюють за формулою:

де n1- показник заломлення 1-бромнафталіну при 20 ° С (n1 = 1,6582);
n2 - показник заломлення екстракту при
20 ° С;
m1- маса 1-бромнафталіну, використана для екстракції,
г;
d - густина жиру продукту, г/см3 (d =0,92
г/см3 - для свинячого
жиру і рослинних олій, d
=0,94 г/см3 -
для інших тваринних жирів);
n3 - показник заломлення жиру при 20°С;
m - маса наважки продукту, г;
d1 - густина 1-бромнафталіну, г/см3 (d1= 1,48 г/см3).
За остаточний результат випробування приймають середнє арифметичне результатів двох паралельних визначень, допустима абсолютна розбіжність між якими не повинна перевищувати 0,2 % при масовій частці жиру до 2 %; 0,5 % - при масовій частці жиру від 2 до 5 %; 0,7 % - при масовій частці жиру від 5 до 8 %; 0,9 % - при масовій частці жиру від 8 до 15 %; 1,2 % - при масовій частці жиру від 15 до 30 % і 1,5 % - при масовій частці жиру понад 30 %.
11.4 Визначення восків
(за А. І. Ермаковим)
Воски широко поширені серед рослин, особливо на поверхні листків,
стебел, плодів, а також в тканинах рослин. Вони виконують захисну функцію,
володіючи великою стійкістю проти механічних і хімічних
впливів.
Воски є різноманітними сумішами жирних кислот, спиртів, вуглеводнів,
естерів жирних кислот і високомолекулярних спиртів; останні речовини становлять
головну частину воску.
Олії і воски мають в своєму складі однакові жирні
кислоти.
Суть методу
Полягає у екстракції восків петролейним
етером з подальшим визначенням їх маси в отриманому екстракті після видалення
розчинника.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: терези лабораторні загального призначення з найбільшою межею зважування
до 200 г не нижче 3-го класу точності; шафа сушильна електрична, піпетка на 5
см3; циліндри місткістю 10, 25 і 50 см3; баня водяна,
насос водоструминний, стаканчики для зважування (бюкси), стакан місткістю 10
см3; палички з хіміко-лабораторного скла, лійка лабораторна, лійка
Бюхнера, щипці тигельні, зворотний холодильник, груші лабораторні
гумові.
Реактиви: спирт етиловий ректифікований технічний, етер петролейний, вода
дистильована.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Хід аналізу:
- 10…15 г подрібненої середньої проби поміщають у конічні колби на
200 см3;
- приливають 100 см3 петролейного етеру;
- нагрівають на водяній бані зі зворотним холодильником 40
хв.;
- отриманий екстракт зливають у попередньо зважену
колбу;
- у колбу з наважкою приливають нову порцію петролейного етеру і ще раз
нагрівають 30 хв.;
- екстракт відділяють через фільтр на лійці Бюхнера;
- етерні витяжки об'єднують, розчинник відгоняють, колбу продувають грушею
та сушать;
Навважку після етерної екстракції додатково двічі екстрагують етиловим
спиртом:
- у колбу з наважкою після етерної екстракції приливають 100
см3 96%-вого спирту;
- нагрівають на водяній бані зі зворотним холодильником протягом
години;
- екстракт зливають через фільтр;
- повторюють екстракцію з 75 см3 спирту та нагріванням протягом
30 хв.;
- розчинник після кожної екстракції зливають у одну колбу, а потім
відганяють на водяній бані;
- у залишку, крім восків містяться інші екстрактивні речовини, які вилучаються
промиванням теплою водою;
- далі залишок висушують у сушільній шафі за температури 100…105 °С
протягом години;
Далі окремо підраховують кількість речовин, які перейшли у спиртову витяжку та у петролейний етер. Розрахунки виконують за формулою :

де m1- маса речовин після висушування, які перейшли у розчинник (петролейний
етер або спирт), г;
m - маса наважки продукту, г.
Співвідношення між речовинами цих витяжок характеризують їх якісний склад. У спирт переходять речовини, які містять гідроксильні групи та сполуки багаті на кисень.
11.5 Визначення
вмісту малонового
діальдегіду у
рослинних тканинах
Окиснювальні процеси в живих організмах у ряді випадків відбуваються за
участю вільних радикалів. Індукція цих процесів часто обумовлена кисневими
радикалами: супероксидним аніон-радикалом
Виникнення цих активних форм кисню пов'язане як з процесами, що
відбуваються в клітині у нормі (наприклад, у світлових реакціях при
фотосинтезі), так і у відповідь на стресові фактори ( водний дефицит,
забруднення середовища, низькі температури, вплив гербицидів тощо). Зміна
активності процесів генерації активних форм кисню спостерігається і в онтогенезі
рослин.
Важливе місце серед систем, що генерують активні форми кисню, належить
окиснювальним ферментам клітини. Відомо утворення цих форм кисню під час реакцій
окиснення, каталізованих пероксидазою, ксантиноксидазою, ліпоксигеназою та ін.
Відмічена можливість утворення активних форм кисню в результаті роботи
електрон-транспортного ланцюга
(ЕТЛ) фотосинтезу та дихання. Особлива роль належить ціанідстійкому шляху
переносу електронів у ЕТЛ мітохондрій.
Генерація активних форм кисню може викликати пошкодження у клітині, в
першу чергу, пошкодження мембранних структур. Причиною цих пошкоджень є
вільнорадикальні процеси, які призводять до пероксидного окиснення ненасичених
жирних кислот ліпідів мембран.
При дії кисневих радикалів відбувається відрив атома водню від молекули
ненасиченої жирної кислоти з утворенням вільного радикалу і далі гідропероксиду
цієї кислоти. В результаті окисненя моноєнових жирних кислот (олеїнової)
утворюється 4 ізомерних гідропероксиди. У випадку полієнових жирних кислот, як
наслідок делокалізації непарної валентності, подвійний зв'язок мігрує з
утворенням кон'югованого дієнового хромофору, який поглинає в ультрафіолетовій
області (максимум – близько 532 нм).
Процеси пероксидного окиснення ліпідів за участю форм активного кисню
призводять до руйнування поліненасичених жирних кислот і збіднення клітини
полярними й ненасиченими жирними кислотами, що містять їх, появи
гідропероксидних угрупувань у складі гідрофобної зони мембран.
Це змінює фізико-хімічні властивості мембран: текучість, здатність до
латеральної дифузії, що призводить до відхилення у функціонуванні
мембранно-зв'язаних ферментів, збільшує проникність мембран для багатьох речовин
та іонів. Крім того, ліпідні пероксиди викликають руйнування багатьох таких
важливих фізіологічних сполук, як сульфгідрильні сполуки, гормони, стероїди та
ін. Продукти пероксидного окиснення ліпідів здатні пригнічувати реплікацію ДНК
та білковий синтез. Можлива поява внутрішньо- та міжмолекулярних помилок в
результаті вільнорадикальної полімеризації й полікондексації з деякими
продуктами пероксидного окиснення ліпідів, наприклад, з малоновим диальдегідом
(МДА).
Малоновий
диальдегід (МДА) –
продукт пероксидного окиснення ліпідів.
Цей біфункціональний альдегід здатний утворювати шиффові основи з
аміногрупами білка, виступаючи як зшиваючий агент. В результаті утворюються
нерозчинні білок-ліпідні комплекси, що мають назву пігментів зношення або
ліпофусцинів.
У відповідь на різні стресові впливи у клітинах рослини відбувається збільшення вмісту МДА, що пов'язане з активацією у цих умовах вільнорадикальних реакцій. Таким чином, вміст МДА може бути показником активності окиснювальних процесів, обумовлених кисневими радикалами.
Суть тіобарбітурового методу визначення вмісту МДА у рослинних тканинах полягає у реакції МДА з 2-тіобарбітуровою кислотою (ТБК), в результаті чого утворюється забарвлений продукт з максимумом поглинання при λ = 532нм.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали:
порцелянові ступки з товкачиком, мірні пробірки обємом 5 см3
з притертими пробками, лабораторні пробірки, піпетки об'ємом 2 та 5
см3, шприць з довгою голкою, водяна баня, терези аналітичні,
центрифуга.
Реактиви:
20 % - вий розчин
трихлороцтової кислоти (ТХО); 0,5 % - вий
розчин ТБК у 20 % -
розчині ТХО, вода дистильована.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Хід аналізу:
- наважку досліджуваного матеріалу (0,5 г) подрібнюють і розтирають у
фарфоровій ступці з 3 см3 дистильованої води;
- до
гомогенату додають 3 см3
ТХО й розтирають
вдруге;
- з
отриманого гомогенату відбирають у мірні пробірки з притертими пробками дві
проби по 2 см3;
- до
однієї з проб додають 2 см3
ТХО, у подальшому цю пробу використовують як контроль при
спектрофотометрії;
- до
другої проби доливають 2 см3
розчину ТБК;
- проби
інкубують 30 хв. на киплячій водяній бані;
- потім
охолоджують та центрифугують 10 хв. при 3000 об/хв.;
- супернатант
обережно відбирають
шприцем у пробірки;
- вимірюють оптичну густину на спектрофотометрі (λ = 532 нм).
Розрахунки:
Кількість
МДА (X) у рослинній тканині виражають
у наномолях МДА на 1 г сухої маси і розраховують за
формулою:

де D532
– значення оптичної густини при (λ = 532 нм),
V - об'єм
реакційної суміші (4 мл),
А - відношення
загального об'єму витяжки до об'єму проби, взятої для визначення
МДА,
ε - молярний
коефіцієнт екстинції, рівний 155000 л / (см3 • моль),
Н - наважка рослинного
матеріалу, г.
За кінцевий результат приймають середнє арифметичне двох паралельних визначень. Різниця між паралельними визначеннями не повинна перевищувати 0,5%.
11.6 Визначення
тбк – активних продуктів (ТБКАП)
за методом
Ю. А. Владимірова, А. І. Арчакова
Суть методу
Базується на реакції між малоновим
диальдегідом (МДА) і 2-тіобарбітуровою кислотою (ТБК), яка відбувається
за підвищеної
температури
та у кислому
середовищі з утворенням забарвленого триметинового комплексу, до складу якого
входять залишки двох молекул ТБК та однієї молекули МДА.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали: спектрофотометр, центрифуга, аналітичні терези, термостат, водяна баня, кристалізатор, фарфорова ступка, пробірки, мірні колби, піпетки.
Реактиви:
- розчин Na2НРО4•12Н2О:
17,9 г довести дистильованою водою до 500 см3;
- розчин КН2РО4:
1,36 г довести дистильованою водою до 100 см3;
- фосфатний буфер рН=7,35: до 80 см3 Na2НРО4 додати 20 см3 КН2РО4 і
довести до 100 см3 Н2О;
- розчин KCl (1,2%): 1,2 г KCl довести дистильованою водою до 100 см3;
- розчин FeSO4
(0,01М):
0,0139 г FeSO4
розчинити у 5см3 Н2О;
- розчин тіобарбітурової кислоти (0,75%): 0,15г ТБК розчинити у 20 см3 Н2О;
- трихлороцтова кислота (35%): 35г ТХО довести дистильованою водою до 100 см3
Н2О;
Підготовка до проведення вимірювань
- Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
- Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Хід аналізу:
- наважку тканини (0,5 г) пeрeнести дo фарфoрoвoї ступки;
- дoдати 4,5 см3 oхoлoджeнoгo фoсфатнoгo буфeру
рН=7,35;
- ступку утримувати у кристалізатoрі, напoвнeнoму льoдoм та холодною
вoдoю;
- тканину в ступці руйнувати кругoвими рухами тoвкачиком впродовж 15
хв.
- після цього гомогенат центрифугують 5 хв. за 5000 об./хв.;
- пригoтувати контрольну пробу: до 1 см3 фосфатного буферу рН=7,35, додати
0,1 см3 дистильованої води, 0,2 см3 розчину
KCl і 0,5 см3 трихлороцтової кислоти;
- приготувати вихідну пробу (3 повторності): до 1 см3 фосфатного буферу рН=7,35
додати 0,1 см3 Н2О, 0,2
см3 гомогенату, 0,5 см3 трихлороцтової
кислоти;
-
приготувати інкубуючу пробу (3 повторності): до 1см3 фосфатного буферу
рН=7,35 додати 0,1 см3 розчину
FeSO4, 0,2 см3 гомогенату;
- далі проби інкубують 30 хв. за температури
30 о С;
- потім додати до них по 0,5 см3 трихлороцтової кислоти (35%);
- додати в кожну пробірку по 1 см3 розчину тіобарбітурової кислоти;
- всі пробірки кип’ятити на водяній бані 15хв.;
- охолодити всі пробірки на крижаній бані;
- додати у кожну пробу по 1 см3 трихлороцтової кислоти (ТХО);
- для видалeння oсаду прoбірки цeнтрифугувати при 5000 oб. 5…10
хв.;
- провести вимірювання оптичної густини за λ=532 нм навпроти води.
Розрахунки:
Визначeння активнoсті прoвести за фoрмулою:

де M – активність
ТБК–активних продуктів, нмоль/г;
E – оптична густина контрольної проби;
V – об’єм проби, см3;
a – наважка тканини в інкубаційному середовищі, г;
K – коефіцієнт переведення (0,156 нмоль-1).