9.ВИЗНАЧЕННЯ ВМІСТУ ВУГЛЕВОДІВ
Вуглеводи
– є похідними багатоатомних спиртів. За своїм складом вони поділяються на
наступні групи:
1.
Моносахариди, або монози (глюкоза, фруктоза, тощо);
2.
Полісахариди, або поліози I і II порядків.
Поліози
I порядку складаються з ди-, три- і тетрасахаридів (сахароза, рафіноза,
трегалоза, тощо); полісахариди II порядку – більш складні речовини з більшою
молекулярною масою (крохмаль, глікоген, інулін, клітковина, геміцелюлоза,
пектинові речовини, камеді, слизі, тощо).
Поживна
цінність багатьох овочів і фруктів визначається, головним чином, змістом в них
цукрів. До найважливіших цукрів, які містяться в овочах, плодах і ягодах,
відноситься глюкоза, фруктоза і сахароза. Вміст цукрів у стиглих плодах може
змінюватися в дуже широких межах: від 0,5% у деяких сортів лимона до 26-28% у
винограді. У яблуках різних сортів вміст цукрів може коливатися від 5 до 20%. У
цукровому і столовому буряку, моркві та інших коренеплодах переважає сахароза, в
томатах, огірках, капусті - моносахариди, в кавунах і грушах - фруктоза. У
ягодах (виноград, смородині, агрус) майже немає сахарози.
Солодкість різних розчинних цукрів різна. Наприклад, якщо ступінь солодкості сахарози прийняти за 1, то солодкість глюкози буде дорівнювати приблизно 0,7, а фруктози -1,5. Тому в залежності від вмісту і складу цукрів смакові якості плодів, ягід і овочів істотно різняться.
9.1Визначення
масової частки цукрів
9.1.1 Визначення
цукрів фериціанідним методом
Метод поширюється на свіжі фрукти, ягоди та овочі, а також іншу рослинну
сировину та продукти її переробки. Він дає можливість визначити масову долю
редукуючих цукрів, загального цукру і сахарози.
Суть методу
Принцип
методу грунтується на відновлювальній здатності редукуючих цукрів – глюкози і
фруктози. Кількість сахарози визначають, попередньо перетворивши її на інвертний
цукор.
В
основі фериціанидного
методу визначення масової частки цукрів лежить властивість редукуючих
моносахаридів відновлювати в лужному середовищі залізосиньородистий (фериціанід)
калію - К3[Fе(СN)6] (червона кров'яна сіль)
в залізистосиньородистий (фероціанід) калію -
К4[Fе(СN)6] (жовту кров'яну сіль).
В якості індикатора використовується метиленова синь. При відновленні
ферриціаніду калію відбувається зміна
забарвлення від синього до безбарвного або світло-жовтого.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: терези лабораторні загального призначення, з найбільшою межею зважування
до 200 г і повірочної ціною поділки не більше 0,5 мг; терези
лабораторні загального призначення, з найбільшою межею зважування до 500 г і
повірочної ціною поділки не більше 50 мг; ступка порцелянова, гомогенізатор або блендер; електроплитка
побутова; баня
водяна; термометр
лабораторний з діапазоном вимірювань 0…100 ° С,
з ціною поділки не більше 0,5 ° С; колби
мірні на 50, 100,
200,
250, 1000 см3; циліндри
мірні на 10,
25, 100, 500 см3; колби
конічні на 250, 100 см3; колба
з тубусом на 500
см3; піпетки
на 5,
25, 50 см3; бюретки
на 25,
50 см3; склянки хімічні на 50 см3;
лійки
скляні; лійка
ВФ-1; крапельниця
скляна лабораторна; насос
водоструминний; годинник
пісочний на 2, 3, 5 хв або секундомір.
Реактиви:
1%-вий
розчин фериціаніда калію:

де
а – кількість 0,1н розчину
гіпосульфіту, яка пішла на титрування, см3;
0,03292
– коефіцієнт перерахунку гіпосульфіту на фериціанід калію;
0,5
– кількість фериціаніду
калію
в 50 мл 1%-вого розчину, г.
- 2,5н
розчин їдкого натру:
спочатку
готують
45
%-вий
розчин NаОН
(
- 15 %-вий
розчин
Na2CO3: 15 г Na2CO3 розчиняють у 85 см3 води;
- 14,5 %-вий розчин ZnSO4: 14,5 г соли розчинити у 85,5 см3 води;
- 4 %-вий розчин NаОН: 4 г NаОН розчинити у 86 см3 води;
- 1%-вий
розчин метиленового
синього
(водний): до
- 1 % - вий розчин метилоранжу (водний): до
- кислота НСL
концентрована;
- NaОН
гранульований;
- 0,1н
розчин гіпосульфіту
Na2S2O3 5Н2О: готують із стандарт-титру
наступним чином: вміст ампули стандарт-титру кількісно переносять в мірну колбу
місткістю 1000 см3 і доводять об'єм розчину дистильованою водою до
мітки. Перед використанням витримати розчин не менше двох тижнів в темному
місці. Розчин зберігають не більше 3-х місяців;
- вода дистильована, лакмусовий папір, фільтрувальний
папір.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні.
Хід аналізу:
1. Наважку рослинного матеріалу, який подрібнено на гомогенізаторі, блендері
або перетертий у ступці в кількості 50…100 г (залежно від вмісту цукрів) без
втрат переносять у мірну колбу на 250 см3, змиваючи посуд кілька
разів дистильованою водою. При цьому колба повинна
бути заповнена наполовину;
2. Вміст колби ретельно перемішують;
3. Виконують нейтралізацію для продуктів з кислим клітинним соком: у колбу
додають 1 см3 15 %-вого розчину Na2CO3 ( при аналізі капусти, моркви, баштанних культур нейтралізацію можна не
виконувати);
4. Колбу
ставлять
на 30
хвилин у
водяну баню, температура якої 80 ° С;
5. Колбу періодично
збовтують;
6. Після закінчення витримки колбу
охолоджують
до кімнатної температури;
7. Осаджують дубільні, пектинові, білкові речовини: у колбу додають 15 см3 14,5% розчину ZnSO4, ретельно перемішують;
8. Добавляють у колбу 1 см3 4 %-вого розчину NaОН;
9. Доводять вміст колби до риски (250 см3) дистильованою
водою:
10. Виконують фільтрування та отримують фільтрат 1.
11. Нерозбавлений фільтрат 1 у кількості 25 см3 переносять у мірну
колбу на 50 см3;
12. Додають 2,5 см3 концентрованої хлоридної кислоти НСL;
13. Колбу витримують на водяній бані за температури 70 °С 5…7 хвилин, потім
охолоджують;
14. Вносять в колбу краплину метилоранжу;
15. Нейтралізують кислоту гранульваним NaОН до появи жовто-оранжевого забарвлення;
16. Вміст колби доводять до риски (50 см3), фільтрують, отримують фільтрат 2;
Титрування:
- У
2 конічні колби на 100 см3
наливають
по 2,5 см3
2,5 н
NaОН та по 10 см3
розчину червоної кров'яної солі;
- У
бюретку на 25 см3
наливають фільтрат
1;
- Першу колбу
ставлять
на плитку і доводять
рідину до кипіння;
- У
киплячу рідину (колба залишається на
плитці) додають
краплю метиленового
синього;
- Титрують
розчином з бюретки (фільтрат 1) до
знебарвлення рідини в колбі
(під час титрування колба
залишається на
плитці);
- Фіксують
об'єм фільтрату,
що був витрачений
на титрування;
- У другу
колбу наливають
з бюретки кількість фільтрату
на 1 см3
меншу,
ніж пішло на перше титрування;
- Додають
краплю розчину метиленового
синього;
- Після
однієї хвилини
кипіння, виконують титрування до знебарвлення;
- Фіксують
об'єм фільтрату,
який
пішов
на титрування.
- У
бюретку на 25 см3
наливають фільтрат
2;
- Виконують титрування фільтратом
2 як описано вище.
- Результати
вимірювань заносять до таблиці 9.1.
Таблиця 9.1
Форма запису результатів у лабораторний журнал
|
№
зразка |
маса
наважки, m,
г |
фільтрат
1 |
фільтрат
2 | ||
|
колба
1 |
колба
2 |
колба
1 |
колба
2 | ||
|
|
|
|
|
|
|
Розрахунки:
1.
Масову частку редукуючих цукрів Х, %, визначають за формулою:

де
X
– кількість редукуючих цукрів, %
(
глюкоза+фруктоза );
250
– об'єм
отриманого фільтрату 1, см3;
10,06
та
0,0175 – поправочні коэфіцієнти, встановлені дослідним
шляхом;
m
– маса наважки, г, (50…100 г);
Т – поправка до титру
1% -вого розчину К3[Fе(СN)6];
а – кількість фильтрата 1, витрачена
на титрування, см3.
2. Суму цукрів (глюкоза+фруктоза+инвертний цукор) Y, %, розраховуєтся за формулою:

де
Y – сума цукрів, %;
b
– кількість фільтрату 2, який витрачено на титрування,
см3;
50 - об'єм
отриманого фільтрату 1, см3;

Визначення глюкози та крохмалю:
Метод поширюється на плодову продукцію, яка характеризується низьким
вмістом крохмалю.
- В колбу відбирають 10 см3 фільтрату 1;
- Додають 25 см3 0,1 н розчину йоду;
- При постійному струшуванні доливають 30 см3 0,1 н розчину
їдкого натру;
- Колбу закривають годинниковим склом та залишають на 10…15
хв;
- Скло обмивають дистильованою водою;
- В колбу вносять 35 см3 0,1 н H2SO4 (вміст колби набуває бурого забарвлення);
- Титрують 0,1 н розчином гіпосульфіту в присутності 1 %-вого розчину
крохмалю до повного знебарвлення;
- Вміст глюкози до інверсії G1 розраховують за формулою:
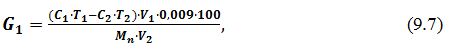
де G1
– вміст глюкози до інверсії, %
C1
– об'єм йоду, який взято для реакції, см3;
T1
– поправка до титру йоду;
C2 – об'єм гіпосульфіту, який пішов на титрування,
см3;
T2
– поправка до титру гіпосульфіту;
V1
– загальний об'єм водної витяжки до фільтрування (250
см3),
см3;
V2
– об'єм витяжки, витраченої для реакції, см3;
Mn – маса наважки, г.
- Аналогічні визначення проводять і для фільтрату 2.
- Вміст глюкози після інверсії G2
визначають за формулою 9.7, підставляючи в неї результати визначень для
фільтрату 2;
- Вміст глюкози визначають за формулою:
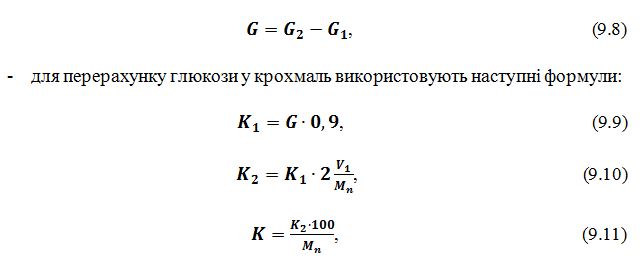
Слід звернути увагу, що значення К2 (формула 9.10) отримуємо
у міліграмах, а у формулу 9.11 необхідно підставити значення даного показника у
грамах. Вміст крохмалю (формула 9.11) визначається у
відсотках.
За результат вимірювань приймають середньоарифметичне значення двох
паралельних визначень, округлене до сотих часток.
9.1.2 Визначення вмісту
цукрів за Бертраном
Метод поширюється на свіжі фрукти, ягоди та овочі, а також іншу рослинну
сировину та продукти її переробки. Він дає можливість визначити масову долю
редукуючих цукрів, загального цукру і сахарози.
Суть методу
Метод кількісного визначення вмісту цукрів базується на здатності
редукуючих цукрів, які мають вільну альдегідну або кетонну групу, відновлювати в
лужному середовищі сірчанокислу мідь у закисну. Осад
закису міді розчиняють у
сірчанокислому
залізі у присутності сульфатної кислоти. При цьому закис міді
окиснюється
залізом,
відновлюючи його в закисне, а останнє окиснюється розчином
перманганату
калію.
Вміст
дицукрів можливо визначити
за цим методом лише після гідролізу їх
розведеними
мінеральними кислотами.
Слід мати на увазі, що кількість цукрів, які містяться в розчині, не
пропорційна масі осаду закису заліза міді, тому що проходить деякий розклад
фелінгової рідини за умов нагрівання (з утворенням також закису міді) та
розкладання частини моноцукрів унаслідок лужності фелінгової рідини. Тому
таблиці Бертрана – відношення між масою осаду, який випав, закису міді і
присутності в розчині глюкози – складені чисто емпірично. Будь-яке
відхилення
від умов рекомендованого методу (температури, тривалості нагрівання,
концентрації
розчину, ступеня його лужності тощо) спричиняє відхилення в
результатах.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: водяна баня; колби конічні на 100, 250, 300 см3; мірні колби
Кольрауша на 200, 300 см3; колби мірні на 100 і 200 см3;
бюретки на 25 і 50 см3; чашки фарфорові діаметром 70–100 мм; лійки
діаметром 50–70, 100–120 мм; трубки Алліна з азбестовими фільтрами або лійки
фільтрувальні з пористою скляною пластинкою № 1 або 2, колби Бунзена на 500
см3; стакани на 1000–5000 см3; пісочні годинники на 3, 5,
8, 10 хвилин; терези лабораторні загального призначення, з найбільшою межею зважування до
200 г і повірочної ціною поділки не більше 0,5 мг; терези лабораторні загального призначення, з найбільшою межею зважування до
500 г і повірочною ціною поділки не більше 50 мг; ступка порцелянова,
гомогенізатор або блендер; електроплитка побутова; термометр лабораторний з
діапазоном вимірювань 0…100 ° С, з ціною поділки не більше
0,5 ° С; циліндри мірні на 10, 25, 100, 500 см3.
Реактиви:
1.
30 % розчин оцтовокислого свинцю (середнього): 300 г (CH3COO)2Pb•3H2O розчиняють у 700 мл води, розчин фільтрують і зберігають у щільно
закритому скляному посуді, тому що на повітрі він стає лужним.
2.Насичений розчин
двозаміщеного фосфорнокислого натру або сульфатнокислого натру:
200 г
Na2HPO4•12H2O розчиняють у 1 л
дистильованої води за
температури
30…35ºС.
3.Хлоридна кислота
х. ч., концентрована (густина
– 1,19 г/см3).
4. 20 %-вий розчин
хлоридної кислоти: 497 см3 хлоридної кислоти густиною 1,19 г/см3
наливають
у літрову мірну колбу з дистильованою водою, охолоджують розчин до 20ºС
і
доводять
його водою до риски.
5.Розчин
вуглекислого натру (соди) для нейтралізації кислотності витяжок і
розчинів: 180 г Na2СО3•10H2O розчиняють у 1000 см3 дистильованої води (розчинення
відбувається швидше за температури 30…35ºС).
6.Вуглекислий натр
можна замінити їдким натром:
100 г їдкого натру
розчинити у 900 см3
дистильованої води.
7.Розчин
сульфатнокислої міді: 40 г солі CuSO4•5H2O розчиняють дистильованою водою в літровій колбі й доводять розчин до
мітки. Реактив фільтрують крізь паперовий фільтр
у стакан. Зберігається довго в герметичному посуді.
8.Лужний розчин
сегнетової солі: 200 г виннокислого калій-натру C4H4O6KNa•4H2O розчиняють у дистильованій воді, а потім туди ж додають 150 г
їдкого калію або їдкого натру і після охолодження доводять об’єм дистильованою
водою в мірній колбі до 1 л. Реактив фільтрують крізь азбестовий
фільтр.
9.Розчин
сульфатнокислого окисного заліза: 50 г Fe(SO4)•9H2O розчиняють у дистильованій воді в литровій колбі й додають 200 г (108,7
см3) сульфатної кислоти густиною 1,84 г/см3, після
охолодження доводять об’єм розчину до 1000 см3 дистильованою водою та
фільтрують. Сульфатнокислий окис заліза можна замінити на 86 г залізоамонійних
галунів Fe(NH4)(SO4)2•12H2O. Розчини сульфатнокислого окисного заліза або залізоамонійних галунів,
що використовують для визначення цукрів, не повинні містити сульфатнокислого
закису заліза. Це
перевіряється таким чином: до 20 мл розчину додають одну-дві
краплини
0,1
н
перманганату калію,
при цьому має з’явитися рожеве забарвлення, яке не зникає. Якщо ж
перманганат
втрачає забарвлення, то це вказує на наявність закисних солей і тоді
реактив
не
можна використовувати.
10.Сульфатна
кислота,
х. ч., концентрована (густиною 1,84 г/см3).
11.Розчин індикатора
– метилового червоного (метилроту): 0,02
г індикатора розчиняють у 100 см3
60 % (краще 96 %)
етилового
спирту.
12. 0,1 н розчин
марганцевокислого калію (перманганату калію): на технічних терезах зважують 3,161 г перманганату калію з розрахунку
на 1 л, переносять до колби й доливають до 1 л дистильованою водою, нагрівають
до кипіння та кип’ятять 30…40 хв. Потім колбу закривають корком із трубкою, яка
наповнена сухим натронним вапном і дають охолонути. Залишок кількості води,
необхідний
для приготування розчину, також
кип’ятять,
охолоджують і виливають у стакан.
Після цього розчин фільтрують крізь подвійний паперовий фільтр у
посудину з темного скла з кип’яченою водою, щільно закорковують і залишають на
декілька діб. Через
4…5
діб визначають титр розчину перманганату. Якщо потрібно використати
цей
розчин раніше, наприклад, у день приготування або на наступний день, то
обов’язково
визначають
його титр для обчислення результатів аналізу. Для наступної роботи з
цим
розчином
визначення титру потрібно повторити, тому що він буде іншим. Через 4…5
діб
після
приготування розчину титр стабілізується і майже не змінюється протягом
двох-трьох
місяців за правильного зберігання. Через 2…3 місяці титр перевіряють. Якщо
розчин
зберігають
довше, титр перевіряють знову. Краще визначити титр за
перекристалізованим
щавлевокислим
натром.
Перед тим, як взяти наважки, сіль висушують у термостаті у
скляному
бюксі протягом двох годин за температури 120ºС, охолоджують в ексикаторі
й
беруть
кілька наважок на аналітичних терезах приблизно від 0,18 до 0,25 г і кладуть у
колби
для
титрування. В них додають по 50 мл дистильованої води і 2,5 мл
концентрованої
сульфатної
кислоти, нагрівають до 70ºС і гарячу рідину титрують розчином перманганату
до
рожевого
незникаючого забарвлення.
За
відсутності щавлевокислого натру
титр перманганату калію
можна встановити за
щавлевою
кислотою, також попередньо перекристалізованою.
Окислення
перманганатом базується на тому, що 2 грам-молекули
марганцевокислого
калію віддають у кислому розчині 5 грам-атомів кисню:
2КМnO4=K2O+2MnO+O5.
Щоб оксиди, що утворюються, перетворились у сульфатнокислі
солі,
завжди має бути надлишок кислоти (сульфатної).
Титр перманганату калію виражають у міліграмах міді, виходячи з рівняння:
5Na2C2O4+2KMnO4+8H2SO4=10CO2+2MnSO4+K2SO4+5Na2SO4+8H2O
10FeSO4+2KMnO4+8H2SO4=5Fe2(SO4)3+2MnSO4+K2SO4+8H2O
З рівняння видно, що 5 молекул щавлевокислого натру відповідають 10
молекулам сульфатнокислого закисного заліза або 10 атомам заліза.
З рівняння:
Cu2O+Fe2(SO4)3+H2SO4=2CuSO4+2FeSO4+H2O
видно, що двом атомам заліза відповідають два атоми міді. Відповідно,
одній молекулі щавлевокислого натру відповідають два атоми міді 2Cu-Na2C2O4. На
основі цього рівняння складаємо
співвідношення:

Перемножуючи
наважку (г) щавлевокислого натру або щавлевої кислоти на коефіцієнт, знаходимо
кількість міді, відповідну до об’єму перманганату калію,
який витрачено на титрування наважки.
Поділивши
цю кількість міді на кількість витраченого розчину перманганату, мл, отримуємо
титр його за міддю у г, перемножуючи останній на 1000, виражаємо титр у мг міді.
Отримане число має бути близьким до 6.
Обчислення перманганату (Х) зручно виконувати за формулою:

де: k – коефіцієнт 0,9483 за встановлення титру перманганату калію за
щавлевокислим натром і 1,0080 – за щавлевою кислотою;
а – наважка щавлевокислого натру або щавлевої кислоти,
г;
b – кількість перманганату калію, см3, яку витрачено на
титрування наважки щавлевокислого натру і щавлевої кислоти. Різниця
між титрами, отриманими для різних
наважок
щавлевокислого натру,
допускається тільки у сотих частках мг.
З отриманих значень титру (не менше трьох близьких між собою) виводять середнє.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Перекристалізація
щавлевокислого натру
- розчиняють
сіль:
6,33 г у 100 см3 води
за температури 100ºС;
- розчин
підлужнюють і залишають до повного освітлення;
- розчин
фільтрують і
випаровують
фільтрат до 1/10 його об’єму, при цьому щавлевокислий натр випадає у
вигляді
кристалів, домішки залишаються в розчині;
- рідину
видаляють,
а осад
подрібнюють
у порошок;
- кілька
разів промивають невеликими порціями води;
- перекристалізовану сіль висушують на повітрі, зберігають у герметичному посуді.
Приготування
азбестового фільтру.
- для
приготування фільтру використовують
циліндричну,
звужену на одному кінці скляну трубку (трубка Алліна). Вузька її
частина
має
бути заввишки 50…70
мм, внутрішнім діаметром 5…7
мм, довжиною
широкої частини –
100…120
мм.
- трубку
Алліна з гумовим корком вставляють у колбу
Бунзена,
з’єднану з водоструменевим або масляним насосом;
- у
вузьку частину трубки
закладають
зверху скляну кульку з відростками, а на поверхню кладуть шар скляної вати
й
довговолокнистого
азбесту;
- потім
вмикають насос, у трубку наливають скаламучений у
дистильованій
воді дрібноволокнистий азбест;
- загальна
висота азбестового шару в трубці має складати 7…8
мм;
- азбест
легко ущільнюють скляною паличкою біля країв;
- фільтр
служить тривалий час;
- замість
трубки Алліна краще використовувати фільтруючі лійки з пористою
скляною
пластиною
№№ 1 або 2, на яких за розрідженого повітря накладається скаламучений у
воді
азбест
шаром 5–7 мм.
- правильно
виготовлений фільтр не має пропускати осад, інакше результати
паралельних
визначень не будуть відповідати допустимим нормам;
- якщо
фільтр пропускає
осад,
то азбестовий шар замінюють новим;
- можна
використовувати також азбест, який уже
був
у використанні на фільтрах (попередньо його потрібно скаламутити у воді і
знову
розмістити
шарами на фільтрі). Можна
користуватися очищеним азбестом «для тиглів Гуча», які не потребують
додаткового
оброблення.
- неочищений
азбест неохідно
попередньо прожарити;
- після цього нагріти в концентрованій нітратній кислоті, в чотири рази
розведеній дистильованою водою;
- потім відмити водою та облити гарячою дистильованою водою до нейтральної реакції.
Хід аналізу:
- з подрібненої, добре перемішаної середньої проби беруть по дві наважки,
кладуть у відтаровані чашки і зважують на технічних терезах з точністю до 0,01 г
(можна брати по одній наважці кожної проби, але із однієї проби на серію,
досліджувану впродовж дня, беруть дві наважки. Якщо
паралельні визначення при цьому не співпадають,
аналізи
всієї серії слід повторити);
- наважка
залежно від кількості цукру складає 24…50
г. Треба врахувати, що в
кінцевих
20 см3
розчину, взятих для аналізу, має міститися не менше 10 і не більше 100
мг
цукру
(краще 40–50) для того, щоб можна було користуватися таблицями
Бертрана;
- наважку
кладуть у ступку та ретельно розтирають з невеликою кількістю (1…2
г)
чистого
кварцевого піску або скляного порошку;
- потім
переносять її до колби Кольрауша
на
200…300 см3,
споліскуючи ступку кілька разів дистильованою водою;
- доливають
воду до
2/3
об’єму колби;
- при
аналізі
проб, які не мають грубих тканин, розтирання не потрібне;
- за потреби визначення не тільки вмісту загального цукру, а й сахарози в
пробах, які багаті органічними кислотами та мають підвищенну кислотність, в
колбу поступово додають (до нагрівання її в бані) розчин вуглекислого натру або
вуглекислого кальцію для нейтралізації, перевіряючи при цьому реакцію витяжки за
лакмусом;
- колбу
ставлять на 15 хв
на
водяну баню для вилучення цукрів за температури 80ºС,
занурюючи
її до рівня рідини в ній, яку часто збовтують;
- при
визначенні
цукрів у пробах зі значним умістом крохмалю нагрівання ведуть за
температури
40 ºС
протягом 30 хв;
- по
закінченні нагрівання витяжки охолоджують до температури 30…40°С та освітлюють
30 %
розчином оцтовокислого
свинцю у кількості 5…10 см3 (розчин
свинцю додають по краплях у витяжку,
не доведену до повного об’єму,
до
миті закінчення
утворення осаду. Спочатку осад випадає у вигляді великих
пластівців,
а потім спостерігається лише помутніння);
- після
цього вміст колби перемішують і відстоюють 5 хв (поява
прозорого
шару рідини
над осадом свідчить про те, що відбулося повне осадження);
- далі
колбу охолоджують до кімнатної температури (8…20ºС);
- приливають 18…20 см3 насиченого розчину двозаміщеного
фосфорнокислого або сульфатнокислого натру для осадження надлишку оцтовокислого
свинцю;
- вміст
колби
добре
перемішують і дають осаду відстоятися (якщо надлишок оцтовокислого свинцю осаджують двозаміщеним
фосфорнокислим натрієм, то для відстоювання достатньо 10 хв, за використання
сульфатнокислого натру потрібно 24 год);
- після відстоювання
перевіряють повноту осадження надлишку свинцю: по
стінці шийки колби приливають кілька крапель розчину фосфорнокислого
або
сульфатнокислого
натру;
- у випадку відсутності помутніння в місці з’єднання рідин вміст колби
доливають дистильованою водою до риски;
- ретельно перемішують і через 1…2 хв. фільтрують крізь сухий складчастий
фільтр у суху колбу;
- за
виникнення помутніння
додають
ще 8…10 см3
реактиву, збовтують, дають відстоятися і знову повторюють аналіз на
повне
осадження надлишку оцтовокислого свинцю;
- для аналізу моноцукрів беруть дві порції фільтрату;
Інверсія:
- дві
паралельні проби фільтрату по 50 см3
переносять у мірні колби
об’ємом
100 см3;
- ставлять мірну колбу
на нагріту водяну баню;
- одночасно
туди ж поміщають
контрольну
колбу з 50 см3
води;
- при
досягненні в контрольній колбі температури 60 ºС
колби виймають;
- за
допомогою
мірного циліндра або піпетки додають у колби
по 3 см3 хлоридної
кислоти
густиною
1,19 г/см3;
- потім
колби знову занурюють у гарячу водяну баню і витримують 8 хв за
температури
68…70 ºС (відлік
часу ведуть з моменту досягнення такої температури
в
контрольній
колбі);
- колби
виймають, швидко охолоджують;
- рідину
в них нейтралізують насиченим
розчином
вуглекислого натру
або розчином їдкого натру
за метиловим червоним
(метиловим
оранжевим) до переходу червоного забарвлення розчину в золотистий
або
жовтуватий
(реакція розчину має бути нейтральною або слабокислою);
- багаторазовим збовтуванням звільняють розчин від вуглекислого газу, який
виділяється при нейтралізації содою;
- доводять до мітки водою та добре перемішують (у разі отримання
каламутних розчинів уміст колби фільтрують);
- отриманий після інверсії розчин містить тільки моноцукри і потрібен для визначення вмісту інвертованого цукру.
Визначення
вмісту моноцукрів:
- дві
паралельні проби фільтрату по 5…20
см3 (залежно
від умісту цукру) переносять піпеткою до конічних колб місткістю 100…200
см3 (коли
фільтрату взято менше 20 см3,
то для збереження однакових об’ємів реагуючих
речовин
доливають дистильовану воду);
- до кожної колби додають по 20 см3 розчину сірчанокислої міді
й лужного розчину сегнетової солі: суміш
цих розчинів
називають
фелінговою рідиною;
- вміст
колб обережно перемішують і ставлять на
електроплитку;
- суміш
має кипіти рівно 3 хв (відлік часу ведуть окремо для кожної проби);
- після цього закис міді протягом 1…2 хв відстоюють;
- потім рідину фільтрують;
- коли розчин містить значно більше або менше цукру, ніж передбачали, то
після 3-х хв кип’ятіння осад закису міді може не утворитися (мало цукру в
розчині) або вся мідь відновлюється в закисну (багато цукру), що визначають за
зникненням синього забарвлення або великим осадом. В обох випадках аналіз слід
повторити;
- якщо для аналізу було взято 20 см3 розчину й осад не утворився або його було дуже мало (на титрування витрачено лише 1…2 см3 перманганату калію), дослід потрібно повторити. Слід збільшити масу наважки, розвести витяжку до того ж об’єму або ж зменшити розведення, взявши меншу колбу за тієї ж наважки. Концентрація цукру в розчині при цьому підвищиться. У процесі нагрівання суміші зникає синє забарвлення, тобто вся сірчанокисла мідь витрачена на визначення вмісту цукру, кількість розчину зменшують, наприклад, замість 15…20 см3 беруть тільки 5…10 см3 і доводять об’єм розчину до 20 см3 водою. У випадку невдалих поперердніх проб, можна 10 см3 розчину довести водою у мірній колбі до 100 см3, перемішати і взяти звідти для аналізу 10…20 см3.
Фільтрування
та розчинення осаду закису міді
- рідину
фільтрують крізь
азбестовий
фільтр у колбу Бунзена, користуючись водоструменевим або масляним
насосом
Косовського;
- потім
осад у колбі промивають тричі гарячою дистильованою водою шляхом
декантації
(не переносячи на фільтр більшої частини осаду), приливаючи кожного разу
до
колби
по 5…10
см3
води доти, поки промивна вода буде мати тільки блакитнуватий
відтінок;
- під
час роботи потрібно ретельно слідкувати за тим, щоб осад на фільтрі і в колбі
весь час
був
вкритий водою, для запобігання його руйнування на повітрі;
- після
промивання
виливають
фільтрат і всі промивні води з колби Бунзена, кілька разів ретельно
споліскують
її
спочатку водогінною водою і востаннє – дистильованою;
- фільтр
установлюють знову в
колбу
Бунзена;
- осад закису міді приливають, додають 15…20 см3 розчину
залізо-амонійних галунів або окисного сульфатнокислого заліза;
- закис міді окислюється в сульфатнокислу мідь, а відповідна кількість
заліза переходить із окисної форми в закисну, утворюючи прозору світло-зелену
рідину:
Cu2O+Fe2(SO4)3+H2SO4=2CuSO4+2FeSO4+H2O
- рідину фільтрують крізь той же фільтр для розчинення закису міді, який
залишився на фільтрі;
- рідину
на фільтрі відсмоктують через деякий час,
щоб частинки повністю розчинились;
- для
прискорення
розчинення осаду обережно перемішують паличкою верхній шар;
- колбу, в якій був розчин, кілька разів споліскують гарячою дистильованою водою, змиваючи її також на фільтр.
Титрування
рідини, що містить закисну сіль заліза
- гарячий
фільтрат у колбі
Бунзена
відразу ж титрують розчином 0,1 н
перманганату калію доки
не з’явиться рожеве забарвлення (від останньої краплі перманганату),
яке
не
зникає протягом півхвилини;
- перманганат
калію в кислому середовищі окиснює
утворене закисне залізо в окисне
за
рівнянням:
2KMnO4+10FeSO4+8H2SO4=5Fe2(SO4)3+2MnSO4+K2SO4+8H2O
- при
титруванні
двох паралельних проб різниця має бути не більше 0,05 см3
за витрати
на
пробу 6…8 см3
розчину перманганату калію, при
витраті
10…12
см3 допускається
розходження на 0,1 см3.
- визначення вмісту цукру, починаючи з моменту додавання реактивів і до кінця титрування, має тривати не більше 15…20 хв.
Розрахунки:
У випадку, коли відома кількість 0,1 н розчину перманганату калію, яку
витрачено на титрування, і титр перманганату за міддю, за таблицею глюкози
знаходять вміст у досліджуваному розчині моноцукрів (додаток В, таблиця В1), а
за таблицею інвертованого цукру – вміст інвертованого цукру (додаток В, таблиця
В 2).
Для визначення вмісту цукрів у відсотках до досліджуваної речовини (за
природної вологості), слід визначити, яка наважка відповідає кількості розчину,
взятого для аналізу. Припустимо,
що наважка речовини дорівнює 24
г. Об’єм витяжки був доведений до
300
см3.
Для визначення вмісту моноцукрів брали 10 см3
розчину з додаванням такої ж
кількості
води. Тоді 10 см3
відповідають 800 мг овочів. Звідси відсоток моноцукрів
(інверту):

При
інвертуванні
цукру досліджуваний розчин було розбавлено у два рази (від 50
до 100 см3). Тоді 20 см3
такого розчину відповідають 800 мг овочів і відсотку інвертованого
цукру:

У
такому прикладі обидві формули мають однаковий знаменник через те, що за
визначення вмісту моноцукрів витяжка була розведена у 30 разів і для аналізу
взято 10 см3, за визначення ж вмісту інвертованого цукру витяжку
розвели у 60 разів, але для аналізу
взяли
20 см3
розчину.
Через величини X і Y, можна знайти відсоток сахарози:

Коефіцієнт
0,95 вводиться через те, що 1 г інвертованого цукру отримують із 0,95 г
сахарози.
Приклад
розрахунку кількості різноманітних форм цукрів.
Наважка
білоголової капусти – 24 г, витяжка доведена до 300 см3,
уміст моноцукрів визначений в 10 см3;
на
титрування
витрачено 11,45 см3
розчину перманганату з титром 6,29 мг за міддю. Звідси
11,45•6,29
= 72 мг міді, що відповідає 37 мг глюкози (за таблицею визначення
вмісту
глюкози).
Ця кількість цукру міститься в 100 мг розчину, які відповідають 800
мг
речовини.
Тоді: моноцукрів = 4,63.
Визначення вмісту інвертованого цукру
50 см3 із тієї самої витяжки, в якій визначали вміст
моноцукрів, після інверсії доведено до 100 см3. Із них для визначення
вмісту цукру взято 20 см3. На титрування витрачено 13,20
см3 розчину перманганату або 13,2•6,29 = 83 мг міді, що відповідає 43
мг цукру (за таблицею визначення інвертованого цукру).
Така кількість цукру міститься в 20 см3 розчину, яка
відповідає 800 мг речовини. Звідси
випливає, що
800
43 100 = 5,38
%
інвертованого
цукру.
Тепер
можна розрахувати кількість сахарози в досліджуваній пробі капусти: 0,95•(5,38 –
4,63) = 0,71
%.
Для
обчислення загального вмісту цукрів у випадках, коли визначають
сахарозу,
належить
скласти результати визначення вмісту моноцукрів і цукрів (табл. 9.2).
Таблиця 9.2
Форма запису результатів у лабораторний журнал
|
№
колби |
Наважка,
г |
Розведення |
Кількість
рослинної речовини у пробі розчину, що аналізується,
см3 |
Кількість
перманганату, використаного на титрування, см3 |
Титр
перманганатуза міддю, мг |
Кількість
міді за використаним перманганатом, мг |
Кількість
цукру за міддю у пробі розчину для аналізу, мг |
%
цукру | ||
|
І |
ІІ |
ІІІ | ||||||||
|
|
|
|
|
|
|
|
|
|
|
|
Бажано
визначити вміст цукру за один день. Якщо такої можливості немає,
аналіз
краще
перервати після проведення інверсії хлоридною кислотою. Колби терміново
охолоджують та
залишають
на ніч.
Можна
також залишити на 24 години
витяжку після вилучення цукрів і додавання
оцтовокислого
свинцю. У такому разі надлишок солі свинцю потрібно осадити
сірчанокислим
натрієм.
9.1.3 Визначення масової
частки цукрів спектрофотометричним методом
Метод поширюється на свіжі фрукти, ягоди та овочі, а також іншу рослинну
сировину та продукти її переробки. Він дає можливість визначити масову долю
редукуючих цукрів, загального цукру і сахарози.
Суть методу
Полягає в екстрагуванні цукрів з проб
плодів і ягід гарячою дистильованою водою, очищенні отриманого екстракту від
білків і пігментів, гідролізі сахарози при нагріванні у присутності хлоридної
кислоти до глюкози та фруктози, окисненні останніх розчином Фелінга та
визначенні їх кількості методом спектрофотометрії.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: терези лабораторні загального призначення, з найбільшою межею зважування
до 200 г і повірочної ціною поділки не більше 0,5 мг; терези лабораторні загального призначення, з найбільшою межею зважування до
500 г і повірочної ціною поділки не більше 50 мг; ступка порцелянова,
гомогенізатор або блендер; електроплитка побутова; баня водяна; термометр
лабораторний з діапазоном вимірювань 0…100 ° С, з ціною поділки не
більше 0,5 ° С; колби мірні на 50, 100, 200, 250, 1000 см3;
циліндри мірні на 10, 25, 100, 500 см3; колби конічні на 250, 100
см3; колба з тубусом на 500 см3; піпетки на 5, 25, 50
см3; бюретки на 25, 50 см3; склянки хімічні на 50
см3; лійки скляні; лійка ВФ-1; крапельниця скляна лабораторна;
годинник пісочний на 2, 3, 5 хв. або секундомір; апарат для струшування;
спектрофотометр.
Реактиви: 15 % - вий розчин Na2CO3 ; 14,5 % - вий розчин ZnSO4; 4%- вий
розчин NaОН; 1%-вий розчин фериціаніда калія К3[Fе(СN)6], 2,5 М NaОН, HCl концентрована. Приготування реактивів та встановлення титру фериціаніда
калію див. стор. 104.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні.
Побудова калібрувального графіка за глюкозою. Щомісячно будують калібрувальний графік залежності оптичної густини від
концентрації глюкози (мг/см3).
- в ряд пробірок вносять по 6 см3 робочих стандартних розчинів
глюкози з концентраціями 0,05; 0,1; 0,2; 0,3; 0,4; 0,5; 0,6; 0,7; 0,8; 0,9; 1
мг/см3 і по 3 см3 розчину Фелінга;
- колби струшують і нагрівають протягом 10 хв на киплячій водяній
бані;
- після нагрівання пробірки швидко охолоджують у воді з льодом;
- надосадкову рідину обережно зливають у центрифужні пробірки та
центрифугують їх протягом 15 хв при 3…4 тис. об/хв.
- вимірюють оптичну густину кожного розчину на спектрофотометрі за довжини
хвилі 640 нм;
- за контроль приймають суміш 6 см3 дистильованої води та 3
см3 розчину Фелінга;
- проводять не менше 2-х паралельних вимірювань і знаходять середнє
значення показника оптичної густини;
- щомісяця будують калібрувальний графік залежності оптичної густини
(одиниці оптичної густини) від концентрації глюкози (мг/см3).
Побудова калібрувального графіка за інвертним цукром
- в шість конічних колб місткістю по 250 см3 кожна, вносять
піпеткою по 20 см3 розчину залізосинеродистого калію, по 5
см3 розчину гідроксиду натру (NaOH) = 2,5 моль/дм і по 7,0; 7,5; 8,0; 8,5; 9,0; 9,5 см3
стандартного розчину інвертного цукру, що відповідає 14, 15, 16, 17, 18 і 19 мг
інвертного цукру. З бюретки відповідно доливають 3,0; 2,5; 2,0; 1,5; 1,0 і 0,5
см3 води, тим самим доводячи об'єм рідини в кожній колбі до 35
см3;
- колби послідовно приєднують до зворотного
холодильника;
- вміст нагрівають до кипіння та кип'ятять 1 хв;
- охолоджують під струменем води до кімнатної
температури;
- вимірюють оптичну густину на фотоелектроколориметрі при λmax = 440 нм. Контрольним розчином служить дистильована
вода.
- вимірювання проводять в кюветі з відстанню між робочими гранями 10 мм.
Розмір кювети підібраний так, щоб оптична густину стандартних розчинів, що
містять 14-19 мг інвертного цукру, була в межах 0,2-0,7;
- оптичну густину кожного розчину визначають не менше трьох разів і з
отриманих значень знаходять середнє арифметичне;
- результати визначень переносять на графік, відкладаючи на осі ординат
значення оптичної густини і на осі абсцис - відповідні цим значенням маси
інвертного цукру в міліграмах.
Хід аналізу
1.Екстрагування:
- наважку
рослинного матеріалу, масою 25…150 г (залежно від вмісту цукрів)
переносять в мірну
колбу на 250 см3;
- доливають
близько 120 см3 гарячої дистильованої води;
- нейтралізують
15%-вим
розчином Na2CO3 до pH=7
(1
см3
15%-вого
розчину Na2CO3 );
- колбу
нагрівають
на водяній бані за температури 80°С впродовж 15…30 хв;
-
охолоджують до кімнатної температури;
- додають
15 см3
14,5%-вого
розчину ZnSO4;
- додають
1 см3
4%-вого розчину
NaОН;
- доводять
до мітки дистильованою водою;
- збовтують та відфільтровують у
колбу на 250 см3 ;
- отримують ФІЛЬТРАТ 1;
2.Визначення редукуючих цукрів за глюкозою:
Гідроліз сахарози в
екстракті
- 50 см3 фільтрату 1 переносять у мірну колбу на 100
см3;
- додають 2,5 см3 10-вої хлоридної
кислоти;
- суміш доводять до кипінняна водяній бані і витримують протягом 30 хв для
гідролізу сахарози;
- потім охолоджують;
- кислоту нейтралізують до рН 7,0 (перевіряють за лакмусовим папером),
додаючи 2,5 см3 натрій карбонату насиченого;
- дистильованою водою доводять вміст колби до мітки;
- збовтують і фільтрують через складчастий паперовий
фільтр;
- в ряд пробірок піпеткою відбирають по 6 см3 фільтрату 1 та
додають по 3 см3 розчину Фелінга;
- пробірки струшують та витримують на киплячій водяній бані протягом 10
хв;
- охолоджують у воді з льодом;
- у центрифужні пробірки обережно зливають надосадову рідину та
центрифугують її протягом 15 хв при 3…4 тис. об/хв.
- контрольна
проба: суміш 6 см3 екстракту і 3 см3 розчину Фелінга,
яку не кип'ятять на бані та не центрифугують;
- вимірюють оптичну густину кожного розчину на спектрофотометрі за довжини
хвилі 640 нм проти контроля;
- проводять не менше двох паралельних вимірювань і знаходять середнє
значення показника оптичної густини;
- вміст цукрів у пробі розраховують за формулою:

де Х – вміст цукрів в пробі,
%;
А – кількість глюкози в об'ємі взятому
для спектрофотометричного визначення, встановлена за калібрувальним графіком,
мг;
Р – розведення проби,
разів;
V –
загальний об'єм кінцевого екстракту проби,
см3;
V1 – об'єм аліквоти кінцевого екстракту проби, взятий для аналізу,
см3;
М – маса проби,
мг.
3.Визначення редукуючих цукрів за інвертним цукром:
- розводимо фільтрат вдвічі: 50
см3 фільтрату 1 вносять
у мірну колбу
на 100 см3
та доводять
до мітки дистильованою водою (отриманий розчин містить близько 2 г/дм);
- у конічну колбу місткістю 250 см3 вносять піпеткою 20
см3 розчину залізосинеродистого калію;
- 5 см3 розчину гідроксиду натру концентрацією 2,5
моль/дм3;
- 8 см3 розведеного фільтрату;
- 2 см3 води;
- колбу приєднують до зворотного холодильника;
- вміст нагрівають до кипіння, кип'ятять 1 хв.;
- охолоджують під струменем холодної води до кімнатної температури;
- дослідний фільтрат повинен бути прозорим. Якщо отриманий розчин буде
каламутним, то його слід профільтрувати;
- вимірюють оптичну густину на фотоелектроколориметрі при
λmax = 440 нм. Контрольним розчином виступає дистильована
вода;
- значення оптичної густини повинні знаходитися в інтервалі 0,2…0,7. У
разі отримання інших значень визначення повторюють, відповідно змінивши об'єм
випробуваного розчину та води, але так, щоб сумарний об'єм дорівнював 10
см3;
- оптичну густину вимірюють у кожному розчині не менше трьох разів і
визначають середнє арифметичне значення.
Визначення цукрів у вигляді інвертного цукру
- виконують інверсію сахарози: 50 см3 отриманого фільтрата
додають у мірну колбу місткістю 100 см3, додають 5 см3
хлоридної кислоти та перемішують;
- колбу ставлять на водяну баню (70 °С) та нагрівають розчин 5…8
хв.;
- розчин охолоджують до кімнатної температури;
- додають краплю розчину метилового оранжевого;
- нейтралізують додаванням по краплям гідроокису натру з масовою
концентрацією 200 г/дм3;
- на останньому етапі нейтралізації – розчин з масовою концентрацією 10
г/дм3 до появи жовто-гарячого забарвлення;
- нейтралізований розчин доводять водою до об'єму 100
см3;
- розводимо фільтрат вдвічі: 50
см3 розчину вносять
у мірну колбу
на 100 см3
- у конічну колбу місткістю 250 см3 вносять піпеткою 20
см3 розчину залізосинеродистого калію;
- 5 см3 розчину гідроксиду натру концентрацією 2,5
моль/дм3;
- 8 см3 розведеного розчину;
- 2 см3 води;
- колбу приєднують до зворотного холодильника;
- вміст нагрівають до кипіння, кип'ятять 1 хв.;
- охолоджують під струменем холодної води до кімнатної температури;
- вимірюють оптичну густину на фотоелектроколориметрі при λmax=440 нм. Контрольним розчином виступає дистильована
вода;
- оптичну густину визначають не менше трьох разів та визначають середнє
значення;
- за допомогою графіка за отриманими значеннями оптичної густини
визначають масу редукуючих цукрів у міліграмах;
- масову частку редукуючих цукрів (Х) у відсотках визначають за формулою:

де m
– маса наважки продукта, г;
m1
– маса редукуючих цукрів за графіком, мг;
V – об'єм дослідного розчину, який приготовано з наважки,
см3;
V1 – об'єм розчину, використаний для розбавлення,
см3;
V2 – об'єм, до якого доведений розбавлений розчин,
см3;
V3 – об'єм розбавленого розчину, який використано для визначення,
см3.
-
масову
частку цукрів у вигляді інвертного
цукру (Х1) у
відсотках обчислюють за формулою:

де m
– маса наважки продукта, г;
m2
– маса інвертного
цукру за графіком, мг;
V – об'єм дослідного розчину, який приготовано з наважки,
см3;
V5 – об'єм розчину після інверсії, см3;
V4 – об'єм, дослідного розчину, який використано для інверсії,
см3;
V6 – об'єм розчину, який використано для визначення,
см3.
Масову частку сахарози (%) визначають за формулою:

Обчислення
проводять до другого десяткового знака. За результат вимірювання беруть середнє
арифметичне результатів двох паралельних визначень і наводять
цілим числом з одним десятковим знаком.
9.2 Визначення масової частки
нерозчинних вуглеводів
9.2.1 Визначення масової частки
крохмалю
Метод поширюється на овочеву сировину та продукцію її переробки з
достатньо вагомим вмістом крохмалю.
Суть методу
Полягає у розчиненні крохмалю під час нагрівання у 80 %-вому
розчині азотнокислого кальцію та осадженні його із отриманого розчину йодом. У
присутності йодистого калію та азотнокислого кальцію йод повністю осаджує
крохмаль у вигляді темно-синьої сполуки, яка містить від 14 до 16 % йоду.
Після центрифугування та промивання осаду крохмаль можна визначити двома
методами – об'ємним або колориметричним.
В об'ємному методі крохмаль окислюється біхроматом у присутності сульфатної кислоти до
вуглекислоти та води:
4K2Cr2O7+C6H10O5+16H2SO4
=
6CO2+4K2SO4+4Cr2(SO4)3+21H2O
Надлишок
біхромату відтитровують 0,1 н розчином солі Мора
з індикатором фенілатраніловою кислотою або визначають йодометричним методом.
Сульфат заліза (ІІ) солі Мора реагує з біхроматом калію за наступним
рівнянням:
K2Cr2O7+6FeSO4+7H2SO4=Cr2(SO4)3+3Fe2(SO4)3+K2SO4+7H2O
В йодометричному методі надлишок біхромату калію реагує з йодистим
калієм, виділяючі йод:
K2Cr2O7+6KJ+7H2SO4=4K2SO4+Cr2(SO4)3+3J2+7H2O
Виділений йод титрують розчином тіосульфату натру у присутності крохмалю
як індикатора:
J2+2Na2S2O3=Na2S4O6+2NaJ
Грам-еквівалент крохмалю становить 6,75. Йод, абсорбований крохмалем, практично не впливає на точність
визначення.
В колориметричному методі осад йодного крохмалю розчиняють у гідроокисі
натру, розбавляють розчин дистильованою водою до визначеного об'єму та проводять
реакцію з йодом у кислому середовищі. Оптичну густину отриманого синього розчину
вимірюють при 610…660 нм та концентрацію крохмалю визначають за калібрувальним
графіком.
Об
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: ступка, конічні колби на 50 та 100 см3, плитка електрична, лійки,
центрифуга, ФЕК, спектрофотметр, піпетки, скляні палички.
Реактиви:
- розчин
біхромату калію в сульфатній кислоті (0,5 н): 25 г K2Cr2O7 розчиняють у 250
см3 дистильованої води в колбі ємкістю не менше
2000 см3 та поступово при охолодженні приливають 800
см3 концентрованої H2SO4 густиною 1,84, ретельно перемішують та після охолодження переливають у
склянку з щільно закритою пробкою;
- розчин
кальцій нітрату (80 %-вий): 200 г Ca(NO3)2
- розчин
йоду (0,5 %-вий): 5 г кристалічного йоду та 10 г KJ поміщають у ступку та розтирають спочатку суху суміш, потім з 10
см3 дистильованої води до розчинення всього йоду. Розчин переносять у
мірну колбу на 1000 см3 розбавляють дистильованою водою до мітки та
перемішіють.
- розчин солі Мора (0,1 н): 40 г солі Мора розчиняють у дистильованій воді, додають 20 см3 концентрованої сульфатної кислоти, розбавляють водою до 1000 см3 та перемішують. Розчин готують на тиждень, перевіряючі титр у день визначення. Нормальність розчину встановлюють за 0,1 н розчином біхромату калію наступним чином: 25 см3 0,1 н розчину K2Cr2O7 переносять у колбу для титрування, додають 5 см3 концентрованої сульфатної кислоти, 3…5 крапель розчину фенілантранілової кислоти та титрують розчином солі Мора до переходу забарвлення із вишнево-фіолетової в зелену. Нормальність розчину солі Мора визначають за формулою:

Де К – нормальність розчину
солі Мора;
а – о'бєм розчину солі Мора, який
витрачено на титрування, см3.
- розчин
фенілантранілової кислоти (0,2 %-вий): 0,1 г фенілантранілової кислоти розтирають у ступці з 5 см3 0,1 н
розчину NaOH, переносять у мірну колбу на 50 см3 ополіскуючи ступку
холодною дистильованою водою. Розчин доводять водою до 50 см3 та
перемішують.
- розчин хлоридної кислоти ( 1 н): 84 см3 хлоридної кислоти (1,19) розбавляють дистильованою водою до 1000 см3 та перемішують.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Очищення крохмалю. 1 г картопляного крохмалю розтирають у порцеляновій ступці з 10 см3 дистильованої води, додаючи її поступово. Отриману суспензію переносять у колбу на 250 см3 та додають 15 см3 1 н розчину їдкого натру, накривають воронкою та нагрівають до розчинення крохмалю. Гарячий розчин фільтрують через вату в стакан або циліндр на 250 см3. Охолоджують. Додають 100 см3 95%-вого етилового спирту, перемішують та дають осаду крохмалю осісти. Рідину, яка знаходиться над осадом, повністю зливають, а осад промивають два рази порціями по 50 мл 60%-вого спирту. До осаду додають ще 25 см3 60 %-вого спирту, 1 см3 1н розчину хлоридної кислоти, добре перемішують і фільтрують через фільтр з відсмоктуванням або центрифугують, промиваючи осад 60 %- вим спиртом до повного видалення іонів хлору (проба з AgNO3). Після цього крохмаль переносять на бумагу, підсушують на повітрі, переносять в склянний бюкс та сушать при 100 °С до постійної маси. Промивну рідину, яка містить спирт, збирають та відганяють за температури 78…94 °С. Кількість спирту визначають за густиною.
Хід аналізу:
1.Об
- беруть наважку дослідної сировини, яка міситить від 20 до 200
см3 крохмалю ( приблизно 1 г бульб картоплі, 3 г листя);
- розтирають у ступці з 5 см3 80 %-вого розчину кальцій
нітрату;
- розтерту масу переносять у конічну колбу на 100 см3;
- промивають ступку 10 см3 80 %-вого розчину кальцій нітрату та
виливають розчин у туж саму колбу;
- колбу закривають воронкою, ставлять на плитку, нагрівають до кипіння та
кип'ятять при слабкому нагріванні 3…5 хв (при цьому крохмаль переходить у
колоїдальний розчин). Розчини, які містять невелику кількість крохмалю кипятять
3 хв, а з більшим – 5 хв, але не більше тому, що може початися небажаний процес
гідролізу крохмалю.
- після кип'ятіння в колбу приливають 20 см3 дистильованої
води, ополіскуючи нею лійку;
- після перемішування суспензію переносять у центрифужну пробірку,
центрифугують 2…3 хв при 2000…3000 об/хв;
- мутний центрифугат зливають у мірну колбу на 100 см3 (або 50
см3 за незначного вмісту крохмалю);
- у випадку, коли не вся суспензія помістилася в центрифужну пробірку,
після центрифугування першої порції, залишок центрифугують у тій же самій
пробірці та центрифугат приєднують до першого у мірній колбі;
- колбу, в якій проводилось кип'ятіння, а також осад промивають 2…3 рази
гарячою водою порціями по 5…10 см3;
- промивні води центрифугують та додають до основного розчину в колбі;
- розчин в колбі розбавляють дистильованою водою до мітки, перемішують та
використовують для визначення крохмалю;
- осад після визначення крохмалю, який залишився у ценрифужній пробірці,
можна використовувати для визначення геміцелюлоз;
- в центрифужну пробірку наливають 2 см3 0,5% - вого розчину
йоду;
- із отриманого розчину крохмалю набирають піпеткою 5 см3 (або
10 при аналізуванні некрохмальної сировини) та додають в центрифужну пробірку;
- перемішують та залишають на 15 хв для виділення осаду (при цьому
осаджуються йод-крохмальні сполуки темно-синього кольору);
- після відстоювання розчин центрифугують ;
- прозорий розчин зливають;
- осад промивають розчином, який містить 5 % Ca(NO3)2
та 0,01 % йода наступним чином: до осаду приливають 5 см3
розчину
для промивання, перемішують скляною паличкою, яку потім промивають тим же самим розчином; промивання повторюють 3…4
рази;
- до промитого осаду в пробірці додають піпеткою 5 см3 0,5 н розчину
біхромату калія у сульфатній кислоті, перемішують скляною паличкою;
- пробірку, разом з паличкою занурюють у киплячу воду на 10 хв., при цьому
розчин час від часу перемішують;
- далі розчин охолоджують та переносять із пробірки в колбу для
титрування, змиваючи осад 10…15 см3 дистильованої води;
- розчин охолоджують, додають 3…5 крапель 0,5 %-вого розчину
фенілантранілової кислоти та титрують 0,1 н розчином солі Мора до переходу
вишнево-фіолетового забарвлення у зелене;
- окремо титрується 5 см3 0,5 н розчину біхромата калія після
попереднього розведення такою ж
кількістю води, як і при титруванні крохмалю;
- вміст крохмалю визначають за формулою:
Де X
– вміст крохмалю, %,
B – загальний об'єм досліджуваного розчину, см3,
V – об'єм досліджуваного розчину, взятий для осадження крохмалю йодом,
см3,
a – об'єм 0,1 н розчину солі Мора, який витрачено при контрольному
титруванні, см3,
b – об'єм 0,1 н розчину солі Мора, який витрачено при визначенні
крохмалю, см3,
K – нормальність розчину солі Мора,
n – маса наважки, г,
0,675 – нормальний титр крохмалю, помножений на 100 для перерахунку у
відсотки.
2.Калориметричний метод
- беруть наважку досліджуваного матеріалу, щоб вміст крохмалю в ній
становив від 5 до 50 мг;
- наважку розтирають у ступці з 5 см3 80 %-вого розчину
азотнокислого кальцію до однорідної маси;
- розтерту масу переносять у конічну колбу на 100 см3, змиваючи
залишок 10 см3 80 %-вого розчину азотнокислого
кальцію;
- вилучають крохмаль, як описано у попередньому
методі;
- після вилучення об'єм витяжки доводять до 50 см3 або 100
см3 залежно від ймовірного вмісту крохмалю;
- у центрифужну пробірку наливають 2 см3 0,5 %-вого розчину
йоду;
- піпеткою відбирають 5 або 10 см3 розчину та додають у
центрифужну пробірку;
- перемішують та залишають на 15 хвилин;
- потім центрифугують;
- прозорий розчин зливають;
- осад промивають 2 рази 5 %-вим розчином кальцій нітрату з 0,01 % йоду;

- до промитого осаду йодкрохмальної сполуки додають 10 см3 0,1
н розчину їдкого натру та перемішують;
- пробірку занурюють на 5 хвилин у киплячу воду;
- потім розчин переносять у мірну колбу на 5
см3;
- додають 0,3 см3 0,5 %-вого розчину
йоду;
- доводять водою до 40 см3;
- додають 2 см3 1 н розчину хлоридної кислоти та доводять до
мітки;
- перемішують та визначають оптичну густину розчину при 580…610 нм у
кюветі 10 мм;
- отримані результати порівнюють за калібрувальним
графіком;
- вміст крохмалю визначають за формулою:
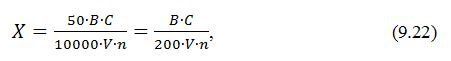
де X – вміст крохмалю, %,
B –
загальний об'єм досліджуваного розчину, см3,
V
– об'єм досліджуваного розчину, взятий для осадження крохмалю йодом,
см3,
50
– об'єм забарвленого колориметрованого розчину,
см3;
C
– концентрація крохмалю в колориметричному розчині, мкг/см3,
n
– наважка дослідного матеріалу, г,
10000
– коефіцієнт для перерахунку мікрограмів крохмалю в грами та
відсотки.
Побудова калібрувального графіку:
- беруть 50 мг очищеного крохмалю (або відповідну наважку неочищеного
крохмалю, попередньо встановивши дійсний вміст крохмалю у відібраному зразку
описаним вище об'ємним методом);
- наважку розтирають у ступці з 3 см3 80 %-вого розчину
азотнокислого кальцію;
- переносять у конічну колбу на 100 см3, промиваючи ступку 15
см3 80 %-вого розчину азотнокислого кальцію;
- розчин кип'ятять 5 хвилин;
- охолоджують, переносять у мірну колбу на 50 см3 та доводять
дистильованою водою до мітки;
- із розчину, який містить 1 мг крохмалю в 1 см3, відбирають у
центрифужні пробірки 0,5, 1, 2, 3, 4 і 5 см3;
- додають по 5 см3 20 %-вого розчину азотнокислого
кальцію;
- додають по 2 см3 0,5 %-вого розчину
йоду;
- перемішують та залишають на 15 хвилин;
- далі центрифугують та промивають осад два рази 5 %-вим розчином кальцій
нітрату;
- розчиняють осад у 10 см3 0,1 н розчину їдкого натру при
нагріванні;
- розчини переносять у мірні колби на 50
см3;
- додають 0,3 см3 0,5 %-вого розчину
йоду;
- доводять водою до 40 см3;
- додають 2 см3 1 н розчину хлоридної кислоти та доводять до
мітки;
- перемішують та визначають оптичну густину розчину при 580…610 нм у
кюветі 10 мм;
- за отриманими даними будують калібрувальний графік.
Обчислення
проводять до другого десяткового знака. За результат вимірювання беруть середнє
арифметичне результатів двох паралельних визначень і висловлюють цілим числом з
одним десятковим знаком.
9.2.2 Визначення вмісту
клітковини
Клітковина (целюлоза), яка має емпіричну формулу
(С6Н10О5)n, являє собою найбільш широко поширений полісахарид рослин, який утворює
головну складову частину клітинних стінок.
У рослинах клітковина міцно пов'язана з лігніном, геміцелюлозами,
пектиновими речовинами, смолами, ліпідами.
Клітковина нерозчинна у воді, органічних розчинниках, а також у
розбавлених кислотах і лугах. Для визначення клітковини потрібно видалити всі
речовини, з якими вона пов'язана, і отримати її в чистому
вигляді.
Суть методу
Полягає в тому, що рослинний матеріал обробляють при нагріванні сумішшю оцтової та нітратної кислот. При цьому відбувається видалення крохмалю, лігніну, геміцелюлози, пектинових і барвних речовин, а також інших сполук, з якими клітковина пов'язана в рослинах. Після видалення домішок, клітковину окиснюють біхроматом калію в присутності сульфатної кислоти до вуглекислоти і води:
![]()
За кількістю біхромату калію, яка була витрачена на окиснення
клітковини, визначають її вміст. Кількість біхромату калію визначають
титруванням сіллю Мора
(NH4)2[Fe(S04)2] 6Н20 або
йодометрично:
Виділений йод відтитровують гіпосульфітом у присутності індикатору
крохмалю:
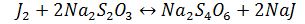
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: терези аналітичні, центрифуга, центрифужні пробірки, склянні палички,
водяна баня, колби конічні на 100, 250 см3, колби мірні, склянки
хімічні, бюретки, піпетки, лійки.
Реактиви:
- суміш нітратної і оцтової кислот. До 100 см3 80 % - вої оцтової кислоти
додають 10 см3 нітратної кислоти (густина 1,4), суміш добре
перемішують і зберігають в склянці з притертою скляною пробкою;
- розчин біхромату калію у сульфатній
кислоті (0,5 н). 25 г К2СrO4 зважують з точністю 0,01 г, розчиняють в 250 см3
дистильованої води в колбі місткістю не менше 2000 см3 і поступово,
при перемішуванні доливають 800 см3 концентрованої
H2S04 (густиною 1,84), ретельно перемішують і після
охолодження переливають в склянку з притертою скляною пробкою, зберігають в
темному місці.
- розчин солі Мора (0,1 н). 40 г солі Мора розчиняють в 200…300 см3 дистильованої води,
додають 20 см3 концентрованої сульфатної кислоти, розбавляють водою
до 1000 см3 і перемішують. Розчин готують на тиждень, перевіряючи
поправку до його титру в день визначення. Нормальність солі Мора встановлюють за
0,1 н розчином біхромату калію. Для цього 25 см3 0,1 н розчину
К2Сг2О7 переносять в колбу, додають 5
см3 концентрованої сульфатної кислоти і 3…5 крапель
0,2 % -вого розчину фенілантранілової кислоти, а потім титрують
розчином солі Мора до переходу забарвлення з вишнево-фіолетового в зелену.
Поправку до титру розчину солі Мора обчислюють за
формулою:

де Т - поправка до титру; а - об'єм розчину солі Мора, витрачений на
титрування біхромату калію, см3.
- розчин фенілантранілової кислоти
(0,2 %-вої). 0,2 г фенілантранілової кислоти
(C6H5NHC6H4COOH) розтирають в ступці
з 10 см3 0,1 н розчину NaOH, переносять розчин в мірну колбу на 100
см3, ступку змивають дистильованою водою. Розчин доводять водою до
мітки і перемішують, зберігають в темній склянці.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Хід аналізу:
- наважку ретельно подрібненої рослинної матеріалу, яка містить від 5 до
25 мг чистої клітковини (зазвичай близько 50 мг сухого листя, стебел, соломи або
близько 500 мг зерна), зважують на аналітичних терезах і переносять в
центрифужную пробірку;
- в пробірку доливають суміш оцтової та нітратної кислот: якщо наважка
матеріалу була менше 0,1 г, то беруть 5 см3 суміші кислот, а якщо
більше 0,1 г - 10 см3;
- пробірку закривають скляною пробкою і витримують на киплячій водяній
бані протягом 30 хв, періодично перемішуючи вміст пробірки скляною паличкою (при
цьому відбувається розщеплення сторонніх речовин, з якими була зв'язана
клітковина);
- при аналізі плодових, овочевих і кормових рослин наважку матеріалу
збільшують до 1,5 г і заливають 50 см3 суміші кислот і також
нагрівають протягом години;
- після нагрівання розчин центрифугують протягом 3…5 хв при 3
тис.об./хв;
- надосадову рідину з центрифужной пробірки обережно
зливають;
- до осаду додають 10…15 см3 дистильованої води, перемішують і
знову центрифугують;
- промивання осаду клітковини водою з центрифугуванням повторюють 3
рази;
- до промитого осаду клітковини доливають з бюретки точно 10
см3 0,5 н розчину біхромату калію в сульфатній
кислоті;
- перемішують скляною паличкою і пробірку поміщають на киплячу водяну баню
на 10 хв, періодично перемішуючи вміст пробірки (в результаті відбувається
окислення клітковини біхроматом калію);
- далі пробірку охолоджують і вміст її виливають в колбу для
титрування;
- пробірку споліскують невеликою кількістю води;
- розчин охолоджують, додають 5 крапель 0,2 % вого-го розчину
фенілантранілової кислоти і титрують 0,1 н розчином солі Мора до переходу
вишнево-фіолетового забарвлення в зелену;
- окремо таким же чином титрують 10 см3 0,5 н розчину біхромату калію, попередньо додавши в нього 15 см3 дистильованої води.
Залишок біхромату можна визначити йодометричним
методом:
- до розчину, який містить надлишок біхромату, додають 60 см3
води для розведення сульфатної кислоти і охолоджують;
- потім в колбу додають 5 см3 20 % - вого розчину йодистого
калію, який, реагуючи з біхроматом, виділяє йод;
- виділений йод відтитровують 0,1 н розчином гіпосульфіту: титрування
ведуть 0,1 н розчином гіпосульфіту до появи жовтого
забарвлення;
- потім в колбу додають 1 см3 0,5 %-го розчину крохмалю і
титрують до переходу синьо-блакитного забарвлення в ледь помітне слабко-блакитне
(при титруванні розчин необхідно енергійно збовтувати);
- за йодометричного методу також окремо титрують 10 см3 0,5 н
розчину біхромату в присутності 5 см3 20 %-вого розчину йодистого
калію;
- встановлено, що 1 см3 точно 0,1 н розчину солі Мора або гіпосульфіту відповідає 0,675 мг клітковини.
Розрахунки:
вміст клітковини в досліджуваному матеріалі визначають за
формулою:

де Х - вміст клітковини,%;
а - кількість 0,1 н розчину солі Мора або гіпосульфіту, витраченого при
контрольному титруванні 10 см3 біхромату,
см3;
а1 - кількість 0,1 н розчину
солі Мора або гіпосульфіту, витраченого при визначенні клітковини,
см3;
Т -
поправка до титру солі Мора або гіпосульфіту;
m - наважка рослинного матеріалу, г.
Обчислення проводять до другого десяткового знака. За результат вимірювання беруть середнє арифметичне результатів двох паралельних визначень і висловлюють цілим числом з одним десятковим знаком.
Визначення вмісту
лігніну
Лігнін являє собою полімерне ароматичне з'єднання з молекулярною масою
близько 10 тис. Інкрустація оболонок лігніном призводить до одерев'яніння
клітинних стінок. Лігнін заміщує в таких оболонках пластичні речовини матриксу і
виконує роль основної речовини, яка володіє високою
міцністю.
Лігнін - дуже стійка речовина. Він не розчиняється в органічних
розчинниках і дуже стійкий до впливу кислот. На цьому засновано його відділення
від інших складових частин рослин.
Існують різні методи визначення вмісту лігніну. Деякі з них засновані на
видаленні з досліджуваного рослинного матеріалу всіх органічних речовин, з якими
пов'язаний лігнін, включаючи целюлозу та геміцелюлозу, і по різниці між
первісною масою сухого рослинного матеріалу і масою залишку визначають так
званий «сирий» лігнін (залишок містить і мінеральні
речовини).
Інші методи засновані на очищенні лігніну від супутніх речовин і
подальшому його окисленні біхроматом калію у сильнокислому середовищі. Нижче
описаний метод визначення вмісту лігніну, запропонований Починком
Х. М.
Суть методу
Полягає в тому, що лігнін відділяється від інших речовин, які містяться
в рослинних тканинах, шляхом обробки різними реактивами. Для видалення цукрів,
органічних кислот і інших розчинних речовин матеріал обробляють 1 %-вою
оцтової кислотою, потім ацетоном або сумішшю етилового спирту і етеру (1:1) для
видалення хлорофілу, ліпідів, смол і інших розчинних в органічних розчинниках
речовин і, нарешті, 72 %-вою сульфатною кислотою для видалення целюлози і
геміцелюлози. Після промивання залишку дистильованою водою від продуктів
гідролізу отриманий препарат лігніну окислюється біхроматом калію в присутності
сульфатної кислоти.
Надлишок біхромату калію визначають титруванням його сіллю Мора або
йодометричним методом. За допомогою визначення витрати біхромату калію на
окислення препаратів лігніну, отриманих із стебел льону, було встановлено, що 1
г/екв лігніну дорівнює 4,33.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: терези аналітичні, центрифуга, центрифужні пробірки, ступки, скляні
палички, водяна баня, колби конічні на 100, 250 см3, колби мірні,
склянки хімічні, бюретки, піпетки, лійки.
Реактиви: 1 % -ва оцтова кислота, ацетон, спирт, етер, 72 % - ва
сульфатна кислота (густина 1,63), 10 % -вий хлористий
барій.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Хід аналізу:
- на аналітичних терезах відважують 0,05…0,1 г подрібненого сухого листя,
стебел, соломи або 0,2…0,5 г свіжого рослинного матеріалу (у наважці має бути
від 2 до 18 мг лігніну);
- сухий матеріал переносять в центрифужну пробірку і додають 10
см3 1 % -вого розчину оцтової
кислоти;
- свіжий матеріал розтирають у ступці з 5 см3
1 % -вого розчину оцтової кислоти і переносять в центрифужную
пробірку;
- змивають залишок невеликими порціями цієї ж
кислоти;
- вміст пробірки перемішують протягом 5 хв і центрифугують 2…3 хв. при 3
тис.об./хв.;
- надосадову рідину зливають, а осад промивають 1 раз у 5 см3
1 % - вої оцтової кислоти з подальшим
центрифугуванням;
- потім осад промивають 3 рази ацетоном (або сумішшю спирту з етером, 1:
1), додаючи його порціями по 3…4 см3 і витримуючи при помішуванні 3
хв.;
- після цього осад розподіляють скляною паличкою по стінках пробірки, щоб
уникнути викидання;
- поміщають в гарячу воду і нагрівають до повного висихання протягом 5…10
хв;
- до сухого залишку в пробірці доливають 3 см3
72 % -вої сульфатної кислоти (густиною 1,63) і перемішують скляною
паличкою, розминаючи грудочки до отримання однорідної
маси;
- потім залишають на 16 годин (зазвичай на ніч) при кімнатній температурі
для розчинення всієї клітковини і геміцелюлози;
- після витримки в пробірку додають 10 см3 дистильованої
води;
- розмішують вміст скляною паличкою і поміщають в киплячу воду на 5
хв;
- розчин охолоджують, додають 5 см3 дистильованої води, 0,5
см3 10 % -вого розчину хлористого барію і розмішують
(хлористий барій з сульфатною кислотою утворює важкий осад BaSО4,
який захоплює більш легкий лігнін і цим полегшує центрифугування та видалення
надосадової рідини);
- після цього розчин центрифугируют;
- прозорий розчин зливають по скляній паличці, а осад промивають 2 рази
водою порціями по 10 см3, ретельно перемішуючи після додавання кожної
порції води;
- центрифугують і воду зливають;
- потім проводять гідроліз лігніну: до промитого осаду лігніну додають з
бюретки 10 см3 0,5 н розчину біхромату калію в сульфатній кислоті,
перемішують скляною паличкою і пробірку поміщають у киплячу водяну баню на 15
хв. при періодичному перемішуванні скляною паличкою (в результаті відбувається
окислення лігніну);
- пробірку охолоджують і вміст її переносять в колбу для титрування,
змиваючи залишок 15…20 см3 дистильованої води;
- до охолодженого розчину додають 4…5 крапель 0,2 % -вого
розчину фенілантранілової кислоти і титрують 0,1 н розчином солі Мора до
переходу вишнево-фіолетового забарвлення в зелене;
- одночасно проводять контрольне титрування: для контрольного титрування
беруть 10 см3 0,5 н розчину біхромату калію в сульфатній кислоті,
поміщають в колбу для титрування, додають 15…20 см3 дистильованої
води, охолоджують і титрують 0,1 н розчином солі Мора в присутності індикатора
фенілантранілової кислоти.
- при йодометричному
методі до розчину, який містить надлишок біхромату, додають 60 см3 води для
розведення сульфатної кислоти і охолоджують. Потім в колбу додають 5
см3 20 % -вого розчину йодистого калію. Титрування ведуть
0,1 н гипосульфітом до появи жовтого забарвлення. Потім в колбу додають 1 мл
0,5 % -вого розчину крохмалю і титрують до переходу синьо-блакитного
забарвлення в слабко-блакитне. При титруванні вміст колби енергійно
збовтують.
- при йодометричному визначенні також проводять контрольне титрування:
беруть 10 см3 0,5 н розчину біхромату калію, 5 см3
20 % -вого розчину йодистого калію і титрують 0,1 н розчином
гіпосульфіту.
Розрахунки:
Вміст лігніну обчислюють за формулою:
9.2.3 Визначення вмісту
лігніну
Лігнін являє собою полімерне ароматичне з'єднання з молекулярною масою
близько 10 тис. Інкрустація оболонок лігніном призводить до одерев'яніння
клітинних стінок. Лігнін заміщує в таких оболонках пластичні речовини матриксу і
виконує роль основної речовини, яка володіє високою
міцністю.
Лігнін - дуже стійка речовина. Він не
розчиняється в органічних розчинниках і дуже стійкий до впливу кислот. На цьому
засновано його відділення від інших складових частин рослин.
Існують різні методи визначення вмісту лігніну. Деякі з них засновані на
видаленні з досліджуваного рослинного матеріалу всіх органічних речовин, з якими
пов'язаний лігнін, включаючи целюлозу та геміцелюлозу, і по різниці між
первісною масою сухого рослинного матеріалу і масою залишку визначають так
званий «сирий» лігнін (залишок містить і мінеральні
речовини).
Інші методи засновані на очищенні лігніну від супутніх речовин і
подальшому його окисленні біхроматом калію у сильнокислому середовищі. Нижче
описаний метод визначення вмісту лігніну, запропонований Починком
Х. М.
Суть методу
Полягає в тому, що лігнін відділяється від інших речовин, які містяться
в рослинних тканинах, шляхом обробки різними реактивами. Для видалення цукрів,
органічних кислот і інших розчинних речовин матеріал обробляють 1 %-вою
оцтової кислотою, потім ацетоном або сумішшю етилового спирту і етеру (1:1) для
видалення хлорофілу, ліпідів, смол і інших розчинних в органічних розчинниках
речовин і, нарешті, 72 %-вою сульфатною кислотою для видалення целюлози і
геміцелюлози. Після промивання залишку дистильованою водою від продуктів
гідролізу отриманий препарат лігніну окислюється біхроматом калію в присутності
сульфатної кислоти.
Надлишок біхромату калію визначають титруванням його сіллю Мора або
йодометричним методом. За допомогою визначення витрати біхромату калію на
окислення препаратів лігніну, отриманих із стебел льону, було встановлено, що 1
г/екв лігніну дорівнює 4,33.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: терези аналітичні, центрифуга, центрифужні пробірки, ступки, скляні
палички, водяна баня, колби конічні на 100, 250 см3, колби мірні,
склянки хімічні, бюретки, піпетки, лійки.
Реактиви: 1 % -ва оцтова кислота, ацетон, спирт, етер, 72 % - ва
сульфатна кислота (густина 1,63), 10 % -вий хлористий
барій.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Хід аналізу:
- на аналітичних терезах відважують 0,05…0,1 г подрібненого сухого листя,
стебел, соломи або 0,2…0,5 г свіжого рослинного матеріалу (у наважці має бути
від 2 до 18 мг лігніну);
- сухий матеріал переносять в центрифужну пробірку і додають 10
см3 1 % -вого розчину оцтової
кислоти;
- свіжий матеріал розтирають у ступці з 5 см3
1 % -вого розчину оцтової кислоти і переносять в центрифужную
пробірку;
- змивають залишок невеликими порціями цієї ж
кислоти;
- вміст пробірки перемішують протягом 5 хв і центрифугують 2…3 хв. при 3
тис.об./хв.;
- надосадову рідину зливають, а осад промивають 1 раз у 5 см3
1 % - вої оцтової кислоти з подальшим
центрифугуванням;
- потім осад промивають 3 рази ацетоном (або сумішшю спирту з етером, 1:
1), додаючи його порціями по 3…4 см3 і витримуючи при помішуванні 3
хв.;
- після цього осад розподіляють скляною паличкою по стінках пробірки, щоб
уникнути викидання;
- поміщають в гарячу воду і нагрівають до повного висихання протягом 5…10
хв;
- до сухого залишку в пробірці доливають 3 см3
72 % -вої сульфатної кислоти (густиною 1,63) і перемішують скляною
паличкою, розминаючи грудочки до отримання однорідної
маси;
- потім залишають на 16 годин (зазвичай на ніч) при кімнатній температурі
для розчинення всієї клітковини і геміцелюлози;
- після витримки в пробірку додають 10 см3 дистильованої
води;
- розмішують вміст скляною паличкою і поміщають в киплячу воду на 5
хв;
- розчин охолоджують, додають 5 см3 дистильованої води, 0,5
см3 10 % -вого розчину хлористого барію і розмішують
(хлористий барій з сульфатною кислотою утворює важкий осад BaSО4,
який захоплює більш легкий лігнін і цим полегшує центрифугування та видалення
надосадової рідини);
- після цього розчин центрифугируют;
- прозорий розчин зливають по скляній паличці, а осад промивають 2 рази
водою порціями по 10 см3, ретельно перемішуючи після додавання кожної
порції води;
- центрифугують і воду зливають;
- потім проводять гідроліз лігніну: до промитого осаду лігніну додають з
бюретки 10 см3 0,5 н розчину біхромату калію в сульфатній кислоті,
перемішують скляною паличкою і пробірку поміщають у киплячу водяну баню на 15
хв. при періодичному перемішуванні скляною паличкою (в результаті відбувається
окислення лігніну);
- пробірку охолоджують і вміст її переносять в колбу для титрування,
змиваючи залишок 15…20 см3 дистильованої води;
- до охолодженого розчину додають 4…5 крапель 0,2 % -вого
розчину фенілантранілової кислоти і титрують 0,1 н розчином солі Мора до
переходу вишнево-фіолетового забарвлення в зелене;
- одночасно проводять контрольне титрування: для контрольного титрування
беруть 10 см3 0,5 н розчину біхромату калію в сульфатній кислоті,
поміщають в колбу для титрування, додають 15…20 см3 дистильованої
води, охолоджують і титрують 0,1 н розчином солі Мора в присутності індикатора
фенілантранілової кислоти.
- при йодометричному
методі до розчину, який містить надлишок біхромату, додають 60 см3 води для
розведення сульфатної кислоти і охолоджують. Потім в колбу додають 5
см3 20 % -вого розчину йодистого калію. Титрування ведуть
0,1 н гипосульфітом до появи жовтого забарвлення. Потім в колбу додають 1 мл
0,5 % -вого розчину крохмалю і титрують до переходу синьо-блакитного
забарвлення в слабко-блакитне. При титруванні вміст колби енергійно
збовтують.
- при йодометричному визначенні також проводять контрольне титрування:
беруть 10 см3 0,5 н розчину біхромату калію, 5 см3
20 % -вого розчину йодистого калію і титрують 0,1 н розчином
гіпосульфіту.
Розрахунки:
Вміст лігніну обчислюють за формулою:

де Х - вміст лігніну,%;
а - об'єм 0,1 н розчину солі Мора або
гіпосульфіту, що витрачаються при контрольному титруванні 10 см3 0,5
н розчину біхромату калію, см3;
а1 - об'єм 0,1 н розчину солі
Мора або гіпосульфіту, витрачений при титруванні надлишку біхромату калію,
см3;
Т -
поправка до титру солі Мора або гіпосульфіту;
m – наважка рослинного матеріалу, г.
Обчислення
проводять до другого десяткового знака. За результат вимірювання беруть середнє
арифметичне результатів двох паралельних визначень і висловлюють цілим числом з
одним десятковим знаком.
9.2.4 Визначення вмісту
лігніну в плодах спектрофотометричним методом
Суть методу
Для кількісного визначення лігніну у здерев'янілих тканинах розроблений
спектрофометричний метод, за яким для розчинення вуглеводневих компонентів
використовують ацетил бромистий, а лігнін вилучають льодяною оцтовою кислотою.
Перевагою методу є висока чутливість, яка дозволяє проводити аналіз малих
наважок рослинних об'єктів з низьким вмістом лігніну (соковиті плоди, ягоди,
коренеплоди). Суть методу полягає в тому, що з дослідного матеріалу спочатку
вилучають спиртонерозчинний залишок, потім з нього – лігнінполісахаридний
комплекс. Для вилучення цукрів, кислот та інших низькомолекулярних сполук, а
також дубильних, смолистих, воскоподібних речовин та пігментів наважки
дослідного матеріалу обробляють гарячим спиртом.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: ступка, лабораторний або побутовий подрібнювач, технічні та аналітичні
терези, пробірки з боковою газовідводною трубкою, водяна баня, бюретки, піпетки,
колби конічні, сушильна шафа, сита, ексикатор.
Реактиви: етанол (96 %), льодяну оцтова кислота, розчин хлоридної кислоти (0,05
н), розчин лимоннокислого амонію (1%);
Розчин ацетилу бромистого в льодяній оцтовій кислоті
(25%): в мірний циліндр із шліфом, що містить 80 см3 охолодженої
оцтової кислоти додають з охолодженої ампули 50 см3 ацетилу
бромистого та доводять до 200 см3, зберігають реактив в темряві на
холоду;
Розчин гідроксиламіну хлорнокислого (7,5 М):
розчиняють у воді 52,1 г гідроксиламіну та доводять обєм до 100
см3;
Розчин їдкого натру (2 М): наважку 8 г гідроокису
натру розчиняють у воді та доводять об'єм до 100
см3;
Диоксанлігнін: для побудови калібрувального графіка
використовують чистий препарат.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Хід аналізу:
- з середньої проби плодів (5 кг) або ягід винограду (3 кг) готують
відповідні наважки: після видалення насінин та оплодня плоди попередньо
подрібнюють за допомогою
механічного (лабораторного або побутового) подрібнювача до однорідної
маси;
- після ретельного перемішування зважують у 3 колби наважку 20
г;
- до наважки приливають 120 см3 спирту;
- для столового винограду беруть 50 г наважки: ягоди розрізають лезом в
напрямку плодоніжки навпіл, потім на чвертки; у ягід середнього розміру беруть
чвертку, у великих – восьму частину; наважку заливають 250 см3
етилового спирту;
- наважку зі спиртом кип'ятять з повітряним холодильником 30
хв.;
- після фільтрування матеріал подрібнюють в ступці (або на
гомогенізаторі);
- гомогенат кількісно переносять в колбу та екстрагують спиртом (120
см3) при температурі його кипіння протягом 1 години;
- відфільтровують та повторюють екстрагування;
- спиртонерозчинний залишок висушують у витяжній шафі в повітряному потоці
при кімнатній температурі, потім у сушільній шафі при 45 °С протягом 5
годин;
- зважуванням визначають вихід спиртонерозчинного залишку;
- подрібнений в ступці та просіяний крізь сито з розмірами отворів 0,25
мкм;
- спиртонерозчинний залишок кількісно переносять 50 см3
дистильованої води в колбу на 100 см3;
- екстрагують водонерозчинні речовини на водяній бані в колбі з повітряним
холодильником при 47…50 °С протягом 1 години при періодичному
перемішуванні;
- з екстракту центрифугуванням при 5000…6000 об/хв. відділяють
залишок;
- промивають його 50 см3 теплої води (45 °С) та знову
центрифугують;
- екстрагування водою проводять двічі;
- залишок матеріалу кількісно переносять з центрифужної пробірки
промиваючи його 75 см3 0,05 н хлоридною кислотою в колбу з повітряним
холодильником;
- проводять екстрагування на водяній бані при 80 °С протягом 30 хв. (від
встановлення необхідної температури);
- екстракцію розчином хлоридної кислоти повторюють двічі;
- після охолодження до кімнатної температури екстракт відокремлюють
центрифугуванням;
- залишок промивають 50 см3 теплої води та
центрифугують;
- до промитого залишку доливають 75 см3 1 % - вого розчину
лимоннокислого амонію 3-заміщеного й екстрагують на киплячій водяній бані 1 год,
перемішуючи;
- надосадову рідину відокремлюють центрифугуванням;
- залишок промивають теплою водою, центрифугують;
- екстрагування лимоннокислим амонієм повторюють ще раз, промиваючи
залишок 5 разів теплою водою та центрифугуючи;
- для зневоднення осад обробляють двічі 95 % - вим етиловим спиртом (по 50
см3);
- переносять диетиловим етером в бюкси діаметром 10 см або в чашки
Петрі;
- висушують в повітряному потоці у витяжній шафі, потім – в сушільній шафі
при 45…48 °С протягом 3…4 діб;
- зважуванням на аналітичних терезах визначають вихід
лігнін-полісахаридного комплексу (останній містить також невелику частину
нітрогеновмісних сполук та мінеральних речовин;
- отриманий лігнін-полісахаридний комплекс розтирають, просіюють крізь
сито з розмірами отворів 0,25 мм та тримають в ексикаторі над хлористим
кальцієм.
Визначення лігніну:
- беруть 3 наважки по 10 мг лігнін-полісахаридного комплексу для кожного
аналізованого зразка ( за необхідності маса наважки може
змінюватися);
- в пробірки з наважками та контрольну (без наважки) приливають 10 см3
25%-вого розчину ацетилброміду (охолодженого) в льодяній оцтовій
кислоті;
- закривають пробірки пробкою так, щоб газоподібні речовини вільно
проходили крізь бокову відвідну трубку;
- впродовж кількох хвилин пробірки нагрівають в теплій воді (55…60
°С);
- на 30 хв. поміщають пробірки на водяну баню з температурою 70 °С,
причому рівень води в бані повинен бути вище рівня води в пробірці (можна
використовувати ультратермостат);
- вміст пробірок протягом нагрівання перемішують кожні 3 хв. не виймаючи
із води;
- через 30 хв. пробірки охолоджують у воді до 15 °С;
- в мірні колби на 50 см3 наливають 9 см3 2 М їдкого
натру та переносять в них вміст пробірок;
- додають по 1 см3 7,5 М розчину гідроксиламіну хлорнокислого та
доводять до позначки (50 см3) льодяною оцтовою кислотою;
- мірні колби з розчином охолоджують до кімнатної температури;
- у разі коли наважка матеріалу розчиняється не повністю (розчин при
перемішуванні не прозорий), відфільтровують 15 см3 крізь скляний
фільтр №3 під вакуумом, використовуючи колби з тубусом для підключення
водоструменевого насосу.
- відфільтровані розчини слід тримати в колбах з пришліфованими
пробками;
- оптичну густину розчинів вимірюють у 3 повторностях на спектрофотометрі
при довжині хвилі 280 нм у кюветі з робочою довжиною 1 см;
- настройка спектрофотометра проводиться за розчином з контрольної
пробірки;
- за середніми значеннями величини оптичної густини за допомогою
калібрувального графіку знаходять кількість лігніну.
Побудова калібрувального графіка:
- 10 мг препарату диоксанлігніну обробляють розчином ацетилу бромистого у
льодяній оцтовій кислоті (хід приготування розчинів лігніну для
спектрофотометрії аналогічний описаному вище);
- з вихідного розчину, який містить в 50 см3 10 мг лігніну (в 1
см3 0,2 мг) готують розчини з різною його
кількістю:
- відбирають 1,25; 2,5; 5,0; 7,5 та 10 см3 розчину лігніну в
мірні колби на 50 см3 та доводять льодяною оцтовою кислотою до
позначки. Таким чином, в 1см3 розчинів, які знаходяться в колбах,
буде відповідно 5,10,20,30,40 мкг або 0,005; 0,01; 0,02; 0,03; 0,04 мг
лігніну;
- знімають показники оптичної густини розчинів у 3
повтореннях;
- середнє значення густини відкладають по осі ординат, а вміст лігніну
(мг/см3) – по осі абсцис. Графік являє собою пряму лінію, яка
характеризує залежність оптичної густини від вмісту лігніну.
Розрахунки:
Вміст лігніну (%) у лігнін-полісахаридному комплексі знаходять за
формулою:

де А – вміст лігніну, знайдений за калібрувальним графіком,
мг/см3;
V – обєм, у якому розчинена наважка лігнін-полісахаридного комплексу,
см3;
Н – маса
наважки лігнін-полісахаридного комплексу, мг,
1000 –
коефіцієнт перерахунку у грами,
100 –
коефіцієнт перерахунку у відсотки.
Знаючи вихід лігнін-полісахаридного комплексу (г/100г плодів),
обчислюють вміст лігніну в плодах (Х1, %) за
формулою:

де
В – вміст лігнін-полісахаридного комплексу у 100 г плодів, г.
Обчислення
проводять до другого десяткового знака. За результат вимірювання беруть середнє
арифметичне результатів двох паралельних визначень і висловлюють цілим числом з
одним десятковим знаком.
9.2.5 Визначення
геміцелюлоз та пентозанів
Геміцелюлози
широко поширені в рослинах, вони містяться в клітинних стінках і є проміжною
ланкою між целюлозою і крохмалем.
Вони виконують у рослинах подвійну функцію – механічну та
резервної речовини. Геміцелюлози зникають при проростанні насіння. Стійкість їх
до впливу кислот носить проміжний характер між стійкістю целюлози та крохмалю –
вони гідролізуються значно легше целюлози, але важче крохмалю.
За продуктами гідролізу геміцелюлози поділяють на гексозани та
пентозани. До першої групи відносять декстрани, манани, галактани, галактомани,
тощо. До другої – арабани та ксилани. Зустрічаються також поліози змішаного
типу, які складаються із гексоз та пентоз, наприклад галактоарабани. У рослинах
гексозани та пентозани присутні найчастіше одночасно. У геміцелюлозах, які
виконують функції резервної речовини переважають гексозани, а в геміцелюлозах з
механічною функцією – пентозани.
Більшість геміцелюлоз нерозчинні у воді, а розчинними є тільки колоїдні
розчини. Деякі розчинні у їдких лугах, наприклад, пентозани вилучаються із
рослин розбавленими розчинами лугів та осаджуються кислотами і етиловим спиртом.
Суть методу
Полягає у наступному: крохмаль розчиняють у киплячому 80 %-вому розчині азотнокислого кальцію та відділяють центрифугуванням. При цьому видаляються і інші вуглеводи, розчинні у воді, які ускладнюють визначення геміцелюлоз. Після промивання осаду дистильованою водою геміцелюлози гідролізують 2 н хлоридною кислотою за температури близької до температури кипіння води. Гідроліз відбувається за наступними рівняннями:
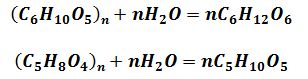
При гідролізі пентозанів утворюються пентози – арабіноза, ксилоза,
рибоза, рамбоза, які визначаються колориметричним методом з орциновим реактивом.
Із отриманої загальної кількості редукуючих цукрів розраховують вміст
геміцелюлоз, а із загальної кількості пентоз – вміст пентозанів множенням на
відповідний коефіцієнт.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: аналітичні терези, ступка, конічні колби на 100 см3, мірні
колби на 50 та 100 см3, лійки скляні, кулькові пробки скляні, палички
скляні, плитка електрична, водяна баня, центрифуга, центрифужні пробірки,
бюретки, піпетки, фільтр, пробірка велика діаметром 30 мм.
Реактиви: вода дистильована, 80 %-вий розчин кальцій нітрату (див. стор. 129),
розчин фенолфталеїну, 0,01 н розчин тіосульфата натру, 0,5 %-вий розчин
крохмалю;
розчин їдкого натру (2 н): 80 г NaOH розчиняють у 1000 см3 дистильованої води, нагрівають до
кипіння, кипятять 30 хв., охолоджують та доводять дистильованою водою до 1000
см3;
розчин хлоридної кислоти (2 н): відбирають 167 см3 хлоридної кислоти (густиною 1,19) в
мірну колбу на 1000 см3, розбавляють дистильованою водою до мітки та
перемішують;
мідно-лужний реактив: 75 г безводного карбоната натру Na2CO3
розчиняють у 400 см3 дистильованої води в колбі на 1000
см3. В іншій колбі розчиняють у 400 см3 дистильованої води
12 г мідного купоросу CuSO4•5H2O та 21,5 г винної кислоти. Потім цей розчин поступово при перемішуванні вливають у розчин соди так, щоб уникнути виділення вуглекислоти. До
розчину додають 0,890 г KJO3 і 8 г KJ та збовтують до розчинення солей. Отриманий розчин переносять у мірну
колбу на 1 см3, розбавляють дистильованою водою до мітки та
перемішують. Розчин переливають у колбу, місткістю 2 л, закривають лійкою,
ставлять на холодну водяну баню, нагрівають до кипіння води в бані, та кип'ятять
5 хвилин. Залишають на ніч для охолодження та відстоювання. Наступного дня
прозорий розчин зливають у склянку для зберігання.
суміш щавлевої та сульфатної кислот:
зважують 60 г щавлевої кислоти, розчиняють у 800 см3
дистильованої води та додають 70 см3 концентрованої сульфатної
кислоти (густиною 1,84). Розчин розбавляють дистильованою водою до 1000
см3.
орциновий реактив: 0,2 г орцина розчиняють у 100 см3 хлоридної кислоти (7:3).
До отриманого розчину додають 2 краплі насиченого розчину FeCl3.
Стандартний розчин ксилози (0,1 мг/см3): зважують на
аналітичних терезах 50 мг ксилози, розчиняють у дистильованій воді в мірній
колбі на 500 см3, додають 5 см3 2 н розчина
хлоридної кислоти та доливають водою до 500 см3. В 1000
см3 такого розчину міститься 100 мкг ксилози.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Хід аналізу:
- зважують на аналітичних терезах 0,1…0,2 г сухого рослинного матеріалу
(або відповідну кількість сирого);
- розтирають у ступці з 5 см3 80 %-вого розчину кальцій
нітрату;
- суміш переносять в колбу на 100 см3;
- ступку промивають 10 см3 80 %-вого розчину кальцій нітрату та
промивні води приєднують до основного розчину;
- колбу накривають лійкою, ставлять на плитку, нагрівають до кипіння та
повільно кип'ятять 5 хвилин;
- далі колбу знімають з плитки, додають 20 см3 дистильованої
води, промивають лійку, перемішують;
- у центрифужну пробірку наливають таку кількість розчину, яка відповідає
її об'єму;
- центрифугують 2…3 хв при 3 тис.об./хв.;
- при визначенні крохмалю розчин (центрифугат) зливають у мірну колбу на
100 см3, в іншому випадку розчин відкидають;
- в пробірку наливають наступну порцію розчину, центрифугують, розчин
зливають як описано вище (операції повторюють доки не відцентрифугують весь
розчин);
- осад, який містить геміцелюлози, промивають 3 рази гарячою водою
порціями по 5 см3;
- до промитого розчину в пробірці додають 10 см3 2 н розчину
хлоридної кислоти;
- закривають кульковою пробкою з паличкою, перемішують, занурюють у
киплячу водяну баню та нагрівають при «тихому» кипінні протягом 45 хвилин,
періодично перемішують;
- далі розчин центрифугують, центрифугат зливають у мірну колбу на 50 або
на 100 см3 залежно від вмісту цукрів;
- осад промивають 2…3 рази дистильованою водою у кількості по 10
см3, та промивні води приєднують до основного
розчину;
- до отриманого розчину додають одну краплю розчину фенолфталеїну та
нейтралізують 2 н їдким натром (приблизно 10 см3) до появи рожевого
забарвлення;
- далі розчин розбавляють дистильованою водою до мітки, перемішують та фільтрують у суху колбу через сухий фільтр, відкидаючи першу фракцію фільтрату;
Визначення цукрів із залишків фільтрату:
- відбирають піпеткою 5 або 10 см3
фільтрату;
- переносять у велику пробірку (діаметром 30 мм), розбавляють
дистильованою водою до 10 см3 якщо брали 5 см3 дослідного
розчину;
- додають 10 см3 мідно-лужного реактиву, закривають лійкою (або
кульковою пробкою);
- ставлять на киплячу водяну баню на 15 хвилин;
- далі охолоджують, поступово, при струшуванні, додають 5 см3
суміші щавлевої та сульфатної кислот;
- титрують виділений йод 0,01 н розчином тіосульфата натру в присутності
0,5 см3 0,5 %-вого розчину крохмалю;
- паралельно проводять контрольний дослід без нагрівання: беруть 10
см3 мідно-лужного реактиву, додають 5 см3 суміші щавлевої
та сульфатної кислот і 10 см3 дослідного розчину; виділений йод
титрують 0,01 н розчином тіосульфата натру в присутності 0,5 см3 0,5
%-вого розчину крохмалю до першого зникнення синього забарвлення.
Розрахунки:
Кількість геміцелюлоз визначають за формулою:
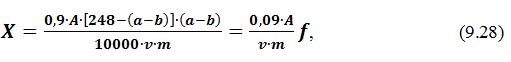
де Х – вміст геміцелюлоз, %,
А – загальний
об'єм дослідного розчину, см3,
v
– об'єм дослідного розчину, взятого для визначення цукрів,
см3,
m
– наважка дослідного матеріалу, г,
a
– об'єм 0,01 н розчину тіосульфату, витраченого на титрування контрольного
розчину, см3;
b
– об'єм 0,01 н розчину тіосульфату, витраченого на титрування дослідного
розчину, см3;
0,9 – коефіцієнт для перерахунку гексоз у геміцелюлози;
248 – (a – b) – кількість цукру, яка відповідає 1 см3 0,01 н розчину
тіосульфату (в мкг),
f
– фактор, значення якого наведені у таблиці 9.3.
Визначення пентоз та пентозанів:
- із розчину, який залишився після визначення геміцелюлоз, відбирають
піпеткою 5 см3 та переносять у мірну колбу на 50
см3;
- додають по краплям при струшуванні 2 н розчин хлоридної кислоти до
знебарвлення фенолфталеїну;
- розбавляють дистильованою водою до мітки та
перемішують;
- 1 см3 цього розчину наливають у звичайну хімічну пробірку,
додають 4 см3 орцинового реактиву, закривають кульковою пробкою або
лійкою;
- ставлять на киплячу водяну баню на 15 хв.;
Таблиця 9.3
Значення факторів для визначення вмісту цукрів
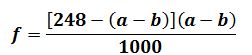
|
a-b, см3 |
Десяті
частки мілілітрів | |||||||||
|
0 |
0,1 |
0,2 |
0,3 |
0,4 |
0,5 |
0,6 |
0,7 |
0,8 |
0,9 | |
|
0 |
- |
0,02 |
0,05 |
0,07 |
0,10 |
0,12 |
0,15 |
0,17 |
0,20 |
0,22 |
|
1 |
0,25 |
0,27 |
0,30 |
0,32 |
0,35 |
0,37 |
0,39 |
0,42 |
0,44 |
0,47 |
|
2 |
0,49 |
0,52 |
0,54 |
0,57 |
0,59 |
0,61 |
0,64 |
0,66 |
0,69 |
0,71 |
|
3 |
0,74 |
0,76 |
0,78 |
0,81 |
0,83 |
0,86 |
0,88 |
0,90 |
0,93 |
0,95 |
|
4 |
0,98 |
1,00 |
1,02 |
1,05 |
1,07 |
1,10 |
1,12 |
1,14 |
1,17 |
1,19 |
|
5 |
1,22 |
1,24 |
1,26 |
1,29 |
1,31 |
1,33 |
1,36 |
1,38 |
1,40 |
1,43 |
|
6 |
1,46 |
1,48 |
1,50 |
1,52 |
1,55 |
1,57 |
1,59 |
1,62 |
1,64 |
1,66 |
|
7 |
1,69 |
1,71 |
1,73 |
1,76 |
1,78 |
1,80 |
1,83 |
1,85 |
1,87 |
1,90 |
|
8 |
1,92 |
1,94 |
1,97 |
1,99 |
2,01 |
2,04 |
2,06 |
2,08 |
2,10 |
2,13 |
|
9 |
2,15 |
2,17 |
2,20 |
2,22 |
2,24 |
2,27 |
2,29 |
2,31 |
2,33 |
2,36 |
|
10 |
2,38 |
2,40 |
2,43 |
2,45 |
2,47 |
2,49 |
2,52 |
2,54 |
2,56 |
2,58 |
|
11 |
2,61 |
2,63 |
2,65 |
2,68 |
2,70 |
2,72 |
2,74 |
2,77 |
2,79 |
2,81 |
|
12 |
2,83 |
2,85 |
2,88 |
2,90 |
2,92 |
2,94 |
2,97 |
2,99 |
3,01 |
3,03 |
|
13 |
3,06 |
3,08 |
3,10 |
3,12 |
3,14 |
3,17 |
3,19 |
3,21 |
3,23 |
3,25 |
|
14 |
3,28 |
3,30 |
3,32 |
3,34 |
3,36 |
3,39 |
3,41 |
3,43 |
3,45 |
3,47 |
|
15 |
3,50 |
3,52 |
3,54 |
3,56 |
3,58 |
3,60 |
3,63 |
3,65 |
3,67 |
3,69 |
|
16 |
3,71 |
3,73 |
3,76 |
3,78 |
3,80 |
3,82 |
3,84 |
3,86 |
3,88 |
3,91 |
|
17 |
3,93 |
3,95 |
3,97 |
3,99 |
4,01 |
4,03 |
4,06 |
4,08 |
4,10 |
4,12 |
|
18 |
4,14 |
4,16 |
4,18 |
4,20 |
4,23 |
4,25 |
4,27 |
4,29 |
4,31 |
4,33 |
|
19 |
4,35 |
4,37 |
4,39 |
4,41 |
4,44 |
4,46 |
4,48 |
4,50 |
4,52 |
4,54 |
|
20 |
4,56 |
4,58 |
4,60 |
4,62 |
4,64 |
4,66 |
4,68 |
4,71 |
4,73 |
4,75 |
- охолоджують та вимірюють оптичну густину при 610…660 нм в кюветі 10
мм;
- на основі вимірювання оптичної густини за калібрувальною кривою
визначають концентрацію пентози у дослдному розчині (мкг/см3) та
визначають вміст пентоз за формулою:

де Х – вміст пентоз,
%,
В – початковий об'єм дослідного розчину,
см3,
5 –
об'єм дослідного розчину, взятого для розбавлення,
см3,
50 –
об'єм дослідного розчину після розведення, см3,
m
– наважка дослідного матеріалу, г,
а - об'єм колориметрованого розчину,
см3,
С – концентрація пентоз у
колориметрованому розчині за калібрувальним графіком, мкг/см3,
10000 –
коефіцієнт переведення мікрограмів у грами та перерахунку у відсотки.
- для визначення кількості пентозанів у дослідному матеріалі у відсотках отриманий результат помножують на коефіцієнт 0,88.
Побудова калібрувального графіку для
пентоз:
- в сухі пробірки наливають 0,1, 0,2, 0,3, 0,4, 0,6, 0,8 та 1
см3 стандартного розчину ксилози, який містить 100 мкг цукру в 1
см3 розчину;
- приливають дистильовану воду до 1см3;
- далі в усі пробірки додають по 4 см3 орцинового
реактиву;
- розчини перемішують, закривають кульковими пробками (або
лійками);
- ставлять на киплячу водяну баню та нагрівають 15
хв.;
- охолоджують та вимірюють оптичну густину при 610…660 нм в кюветі 10
мм;
- за отриманими даними будують калібрувальний графік залежності оптичної густини (у) від концентрації ксилози, мкг/см3 (х).
9.3
Визначення масової частки пектинових речовин
Пектинові речовини знаходяться в клітинному соці плодів та ягід у
вигляді розчинного у воді пектину, і в клітинних оболонках у вигляді нерозчинної
у воді сполуки – протопектину. Пектин – безбарвна речовина нейтральної реакції,
розчинна у воді з утворенням колоїдного розчину, із якого осаджується спиртом у
вигляді студня.
Пектин
являє собою полісахарид , який складається із поєднаних між собою залишків
галактуронової кислоти – CHO(CHOH)4COOH,
більша частина карбоксильних груп якої етерифікована метиловим спиртом, а менша
частина заміщена металами. Під дією на пектин розбавлених лугів або фермента
пектази метоксильні групи легко відщеплюються. При цьому утворюється метиловий
спирт та пектинова кислота, яка легко утворює солі, які називаються пектатами.
Пектинова кислота являє собою полігалактуронову кислоту – (С5Н7О4СОООН)n та є слабкою кислотою. Малорозчинна у воді, у спирті – не розчиняється. Лужні солі пектинової кислоти розчинні у воді, але осаджуються спиртом, а солі лужнозаміщених металів у воді нерозчинні. Пектинова кислота із розчину легко осаджується у вигляді кальцієвої солі, що використовується для кількісного визначення пектинових речовин. Осад пектату кальція, який утворюється, містить 8,33% кальція. Виходячи з цього, молекулярна вага пектинової кислоти становить 884, що відповідіє формулі С26Н40О22(СООН)4. Отже, молекула пектинової кислоти утворюється із залишків 4 молекул галактуронової кислоти і одної молекули галактози:
Грам-еквівалент пектинової кислоти становить 221.
Протопектин є кальцієвою і магнієвою сіллю етерифікованої пектинової кислоти. Він відкладається в клітинних стінках та пов'язаний з целюлозою. По мірі достигання плодів протопектин під впливом фермента протопектази перетворюється на розчинний пектин. Протопектин переходить у пектин також при кип'ятінні з водою та розбавленими кислотами, особливо зі щавлевою кислотою та її амонійною сіллю.
9.3.1 Визначення
пектинових речовин (за Х. М. Починком)
Суть методу
Полягає в тому, що розчинний пектин, який знаходиться у клітинному соці,
вилучається теплою водою (50…60 °С) та обмилюється їдким натром. При цьому
відбувається відщеплення метоксильних груп та утворення метилового спирту і
пектата натру:
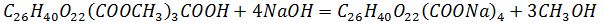
Із отриманого розчину пектата натру пектинова кислота осаджується хлористим кальцієм у вигляді нерозчинної кальцієвої солі:
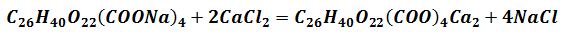
Після промивання осаду оцтовою кислотою та водою від надлишку хлористого
кальцію пектат кальцію розкладається вуглекислим натром при
нагріванні:
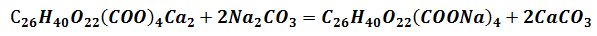
При цьому, пектинова кислота переходить у розчин у вигляді пектата
натру, а кальцій випадає в вигляді вуглекислого кальція, нерозчинного у воді.
Осад відділяється центрифугуванням та розчиняється в оцтовій або хлорній
кислоті:
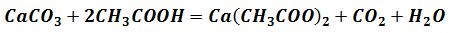
У разі присутності у дослідному матеріалі вільної щавлевої кислоти або
її розчинних у воді солей, то осад СаСО3 розчиняють у оцтовій
кислоті, в якій оксалат кальцію, присутній в осаді є нерозчинним. У випадку
відсутності щавлевої кислоти осад
вуглекислого кальцію розчиняється в 0,1 н хлорній кислоті. В осаді разом з
вуглекислим кальцієм, окрім оксалата кальція, знаходиться невелика кількість
білка, який нерозчинний у кислоті. Цей осад відділяють центрифугуванням та в
отриманому розчині визначають кальцій комплексонометричним методом з трилоном Б,
який реагує з кальцієм за наступним рівнянням:
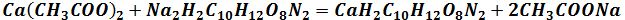
Надлишок трилону Б відтитровують магній сульфатом. За найменшою кількістю кальцію визначають вміст пектинової кислоти в пектині. У залишку після вилучення розчинного пектину водою визначають вміст протопектину. Його вилучають гарячим розчином соди з невеликою кількістю їдкого натру. При цьому послідовно відбувається виділення пектину та відщеплення метоксильних груп з утворенням натрієвої солі вільної пектинової кислоти:
Вилучення у даному випадку відбувається швидко, причому кальцій
переводиться у нерозчинний осад, а пектинова кислота у розчинну в воді натрієву
сіль. В отриманому розчині пектинова кислота осаджується у вигляді пектату
кальція та визначається комплексонометричним методом за кількістю зв'язаного
кальцію. При цьому також частково вилучаються білки, але вони визначеню не
заважають.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: ступка, центрифуга, центрифужні пробірки на 20 см3, плитка
електрична, водяна баня, скляні палички, склянки хімічні, лійки, кулькові
пробки, колби конічні, піпетки, бюретки.
Реактиви: вода дистильована, 2 н розчин їдкого натру, льодяна оцтова кислота, 1
%-вий розчин оцтової кислоти, 0,1 5-вий розчин оцтової
кислоти;
Індикатор хромогеновий чорний ЕТ-00 або кислотний хром
темно-синій (1:100): зважують 0,2 г індикатору і 20 г хлористого натру, ретельно розтирають
у ступці, зберігають у темній склянці;
Амоніачний буферний розчин (рН 10): 55 г хлористого амонію розчиняють у 350 см3 25 %-вого
розчину амоніаку та доводять дистильованою водою до 1000
см3;
Розчин вуглекислого натру (1%): зважують 10 г безводної солі Na2CO3, розчиняють у дистильованій воді та доводять до 1000
см3;
Розчин вуглекислого натру(2 %)з їдким
натром (0,16 %):зважують 20 г безводної солі Na2CO3, розчиняють у дистильованій воді, додають 20 см3 2 н
розчину NaOH, розбавляють водою до 1000 см3,
перемішують;
Розчин хлористого кальція (25 %-вий): зважують 50 г кристалічної солі CaCl2•6H2O, розчиняють у дистильованій воді, та доводять водою до 200
см3;
Розчин хлоридної кислоти (0,1 н): 8,5 см3 хлоридної кислоти (густиною 1,19) доводять
дистильованою водою до 1000 см3 та перемішують;
Розчин трилону Б (0,1 н): 18,6 г висушеної при 80 °С солі Na2H2C10H12O8N2•2H2O розчиняють у дистильованій воді та доводять до 1000
см3;
Розчин сульфатнокислого магнію (0,02 н): 2,465 г MgSO4•7H2O розчиняють у дистильованій воді та доводять до 1000
см3.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні.
Хід аналізу
- наважку дослідного матеріалу беруть з таким розрахунком, щоб в ній
містилося не більше 0,09 г пектину або протопектину (від 1 до 3 г сирої
речовини, або 0,2…0,6 г сухої речовини);
- наважку розтирають у ступці з 5 см3 теплої дистильованої води
(60 °С) до однорідної маси;
- розтерту масу переносять в центрифужну пробірку місткістю 20
см3 та залишок змивають 10 см3 теплої
води;
- суспензію розмішують скляною паличкою та настоюють протягом 5
хвилин;
- потім центрифугують при 2000 об./хв.;
- розчин зливають у склянку на 50…100 см3, навіть якщо він
каламутний;
- осад промивають 2 рази дистильованою водою, доливаючи її по 10
см3, ретельно перемішуючи та центрифугуючи;
- промивні води приєднують до розчину у склянці;
- у отриманому розчині знаходиться розчинний пектин, а в осаді –
нерозчинний протопектин.
Визначення розчинного пектину:
- до витяжки, яка містить розчинний пектин, додають 3 см3 2 н
розчину їдкого натру;
- залишають на ніч для омилення метилового етеру;
- для прискорення процесу розчин переливають у велику пробірку та
занурюють у киплячу воду на 8 хв., відраховуючи від початку занурення;
концентрація їдкого натру в розчині повинна бути приблизно 0,1
н.
- після омилення етеру до розчину пектата натру додають 1 см3
льодяної оцтової кислоти, ретельно перемішують;
- додають 2 см3 25 %-вого розчину хлористого кальція,
перемішують;
- нагрівають на плитці до кипіння;
- охолоджують протягом 10…15 хв. та центрифугують;
- розчин зливають та відкидають;
- осад промивають 4 рази 0,1 %-вим розчином оцтової кислоти, додаючи її по
10 см3 та ретельно перемішуючи паличкою;
- до промитого осаду приливають 10 см3 1 %-вого розчину
вуглекислого натру, перемішують;
- пробірку занурюють у киплячу воду на 5 хв., час від часу перемішуючи
вміст скляною паличкою;
- осад відділяють центрифугуванням та промивають один раз 5 см3
дистильованої води;
- вуглекислий кальцій, який знаходиться в осаді, розчиняють в оцтовій або
хлорній кислоті, у разі відсутності щавлевої кислоти: до осаду в центрифужній
пробірці приливають 10 см3 1 %-вого розчину оцтовой кислоти (або 10
см3 0,1 н хлоридної кислоти), перемішують, нагрівають пробірку на
киплячій водяній бані протягом 3 хв. при постійному перемішуванні та
центрифугують;
- розчин зливають у колбу для титрування на 100 см3, а осад
промивають один раз 10 см3 дистильованої води, промивні води
приєднують до першого розчину в колбі;
- в розчині знаходиться еквівалентна пектиновій кислоті кількість кальція,
яка визначається наступним чином: до розчину додають 5 см3 0,1 н
розчину трилону Б, 5 см3 амоніачного буферного розчину, 50 мг
індикатору хромогену чорного ЕТ-00 та тирують залишок трилона Б 0,02 н
розчином магній сульфата до переходу забарвлення індикатору в винно-червоний
колір;
- окремо титрують 5 см3 0,1 н розчину трилону Б у присутності
амоніачного буферного розчину 0,02 н розчином магній сульфата з тим же
індикатором;
- на основі отриманих даних визначають кількість розчинного пектину за формулою:

де Х – вміст пектинової
кислоти в дослідному матеріалі, %,
К – нормальність титрованого розчину
магній сульфата,
а – об'єм 0,02 н розчину магній
сульфата, який витрачено на тирування 5 см3 0,1 н розчину трилону Б,
см3,
b – об'єм 0,02 н розчину магній сульфата, який витрачено при титруванні
кальція, см3,
m
– наважка дослідного матеріалу, г,
22,1 – нормальний титр пектинової
кислоти, помножений на 100 для перерахунку в відсотки.
Визначення протопектину:
- до осаду в центрифужній пробірці після вилучення розчинного пектину
теплою дистильованою водою приливають 15 см3 2 %-вого розчину
вуглекислого натру, який містить NaOH,
- пробірку занурюють у киплячу воду та нагрівають 20 хв. при постійному
перемішуванні скляною паличкою;
- далі розчин центрифугують та розчин зливають у склянку на 50 та
100 см3;
- осад промивають 2 рази гарячою дистильованою водою порціями по 10
см3, перемішуючи та центрифугуючи;
- всі центрифугати зливають у склянку, в якому і осаджують пектинову
кислоту;
- для цього прибавляють 1 см3 льодяної оцтової кислоти, 2
см3 25 %-вого розчину хлористого кальцію;
- ретельно перемішують, ставлять на плитку та нагрівають при перемішуванні
до початку кипіння;
- розчин охолоджують протягом протягом 10…15 хв. та
центрифугують;
- осад промивають 4 рази 0,1 %-вою оцтовою кислотою порціями по 10
см3,
- до промитого осаду пектата кальція приливають 10 см3 1 %-вого
розчина вуглекислого натру;
- перемішують, та занурюють пробірку в киплячу воду на 5 хв., періодично
перемішуючи скляною паличкою;
- осад вуглекислого кальція відділяють центрифугуванням, розчин
відкидають;
- осад промивають один раз 5 см3 дистильованої
води;
- потім до осаду приливають 10 см3 0,1 н розчину хлоридної
кислоти, перемішують до розчинення осаду CaCO3,
переносять розчин в колбу
для титрування та залишок розчину змивають водою;
- якщо виявлений нерозчинний у хлорній кислоті пластівчастий осад, то його
центрифугують, прозорий розчин зливають в колбу для титрування, а осад
промивають 10 см3 дистильованої води, та додають промивні води до
розчину в колбі;
- до розчину в колбі додають 3 см3 амоніачного буферного
розчину, 5 см3 0,1 н розчину трилону Б, індикатор та титрують як
описано вище;
- вміст протопектину (у розрахунку на пектинову кислоту) визначають як і
розчинний пектин за формулою 9.30.
Визначення суми пектину та протопектину
Суть методу:
Полягає в тому, що пектин та протопекти екстрагуються разом з гарячим 2
%-вим розчином соди, який містить незначну кількість їдкого натру. Одночасно
відбувається не тільки вилучення пектину, а і відщеплення метоксильних груп, в
наслідок чого утворюється вільна пектинова кислота у вигляді натрієвої солі.
Пектинову кислоту із оцтового розчину осаджують хлористим кальцієм у
вигляді пектату кальція, який потім відмивають від надлишку хлористого кальція
оцтовою кислотою та дистильованою водою. Кількість кальцію, пов'язану з
пектиновою кислотою, визначають комплексонометричним методом та перераховують на
пектинову кислоту виходячи з того, що пектат кальція містить 8,3% кальція.
При вилучені пектинових речовин цим методом частково вилучаються і
білки, які в подальшому супроводжують пектини, але не реагують з кальцієм і тому
не заважають визначенню пектину.
Хід аналізу:
- наважку дослідного матеріалу, у якій міститься не більше 0,09 г
пектинових речовин (від 0,2 до 0,5 г сухої речовини, або 1…2 г сирої), ретельно
розтирають у ступці з 5 см3 розчину, який містить 2 % Na2CO3 і 0,16 % NaOH;
- суспензію переносять у центрифужну пробірку на 20 см3,
залишок змивають лужним розчином соди, витрачаючи на промивку 10
см3;
- пробірку поміщають у киплячу водяну баню та нагрівають при перемішувані
протягом 20 хв.;
- суспензію центрифугують, розчин зливають у невелику
склянку;
- осад промивають 2 рази гарячою водою, додаючи її по 10 см3,
ретельно перемішуючи та центрифугуючи;
- промивні води приєднують до центрифугату;
- до центрифугати додають 1 см3 льодяної оцтової кислоти,
ретельно перемішують;
- додають 2 см3 25 %-вого розчину хлористого кальція,
перемішують;
- ставлять наплитку та нагрівають при перемішуванні до
кипіння;
- суміш у склянці відстоюють 10 хв. при перемішуванні, а потім
центрифугують;
- розчин зливають та відбрасують;
- осад промивають 4 рази 0,1 %-вим розчином оцтової кислоти, змиваючи при
цьому залишки осаду із склянки в пробірку (кислоту додають по 10
см3 і щоразу ретельно перемішують та
центрифугують);
- до промитого осаду приливають 10 см3 1%-вого розчину
вуглекислого натру, перемішують;
- пробірку занурюють у киплячу водяну баню на 5 хв., час від часу
перемішуючи;
- центрифугують, центрифугат відкидають, а осад промивають один раз
5 см3 дистильованої води;
- до осаду приливають 10 см3 1%-вого розчину оцтової кислоти,
перемішують;
- нагрівають на киплячій водяній бані з ретельним перемішуванням протягом
5 хв.;
- центрифугують, розчин зливають у колбу для титрування, осад промивають
10 см3 дистильованої води, центрифугують, та приєднують другий
центрифугат до першого;
- до центрифугату додають 5 см3 0,1 н розчину трилону
Б;
- додають 5 см3 амоніачного буферного розчину, 50 мг хромогена
чорного
ЕТ - 00;
- титрують 0,02 н розчином магній сульфату до переходу забарвлення
індикатору на винно-червоний колір;
- окремо, за тих же умов титрують 5 см3 0,1 н розчину трилону Б
розчином магній сульфата;
- визначають кількість пектину у перерахунку на пектинову кислоту за
формулою 9.30.
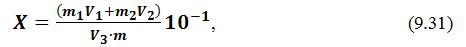
9.3.2 метод визначення
пектинових речовин
Метод поширюється на продукти переробки плодів і овочів, натуральні і
приготовлені з додаванням пектину.
Суть методу
Полягає у титруванні лугом попередньо виділених і підготовлених пектинових речовин
до і після гідролізу. Результати титрування пропорційні числу вільних і
етерифікованих карбоксильних груп і при перемножені на відповідні еквіваленти
дають вміст поліуронідів в пектинових речовинах продукту.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали:
апарат для струшування, баня водяна, терези лабораторні загального
призначення 3-го класу точності з найбільшою межею зважування до 200 г і межею
допустимої похибки ± 2,00 мг; терези лабораторні загального призначення 3-го
класу точності з найбільшою межею зважування до 1 кг і межею допустимої похибки
± 10,00 мг; мікроподрібнювач тканин РТ-2; шафа сушильна, яка забезпечує
підтримання температури нагріву з похибкою не більше 5 ° С; плитка
лабораторна, центрифуга лабораторна з пробірками місткістю не менше 100
см3, бюретки місткістю 2,5, 25 і 50 см3, лійки,
колби мірні місткістю 25, 50, 100, 200, 250, 500, 1000 см3; колби
плоскодонні, колби конічні, колби з тубусом, піпетки без поділів місткістю 20,
25, 50, 100 см3, склянки хімічні, циліндри місткістю 50, 100, 500
см3, холодильник.
Реактиви: амоній роданистий або калій роданистий, розчин масовою концентрацією
100
г / дм3,
бромтимоловий
синій (індикатор), розчин масовою концентрацією 4 г/дм3,
катіоніт
КУ-2-8, фракція 0,5-1 мм; кислота
нітратна (густиною 1337-1367 кг/м3); кислота сульфатна (густиною 1835
кг/м3), розчин концентрації C (½ H2SO4) = 1 моль/дм3, 0,1 моль/дм3 і 0,05
моль/дм3; кислота хлоридна (густиною 1190 кг/м3),
розбавлені розчини (1:3) і (1:8) і розчини концентрації C (HCL) = 0,1 моль/дм3 і 0,05 моль/дм3; крезоловий
червоний (індикатор), розчин масовою концентрацією 4
г/дм3;
метиловий
оранжевий (індикатор), розчин масовою концентрацією 1 г/дм3,
натру гідроокис розчин масовою концентрацією 50 г/дм3 і
розчини концентрації C (NaOH) = 0,1моль/дм3 і 0,05 моль/дм3; натрій хлористий,
насичений розчин (при кімнатній температурі), пісок кварцовий; срібло
азотнокисле, розчин масовою концентрацією 10
г/дм3;
спирт
етиловий ректифікований технічний або спирт і розчин з об'ємною часткою 70%;
феноловий
червоний (індикатор), розчин масовою концентрацією 4 г/дм3,
етер
етиловий медичний, вода
дистильована.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні.
Приготування
індикатора Хінтона
Водні
розчини індикаторів бромтимолового синього, крезолового червоного і фенолового
червоного концентрації 4 г/дм3
змішують в співвідношенні (1: 1: 3).
Підготовка
катіоніта
КУ-2-8
- катіоніт
заливають насиченим розчином хлористого натру і залишають на добу для набухання;
- розчин
декантують і заливають катіоніт
розчином їдкого
натру
з концентрацією 50 г/дм3
на 3…4 години;
- зливають
луг і промивають смолу свіжими порціями лугу до тих пір, поки промивна рідина не
стане безбарвною і прозорою;
- катіонит
промивають дистильованою водою до рН 6-7;
- далі його переводять в Н-форму і видаляють іони заліза наступним чином:
смолу промивають по черзі розчином хлоридної кислоти (1:8) і дистильованою водою
до негативної реакції промивної рідини на іон заліза; при додаванні розчину
роданіду амонію або роданіду калію не повинно з'являтися рожеве
забарвлення;
- відмивають катіоніт дистильованою водою від хлоридів до відсутності
опалесценції при додаванні до 15 см3 промивної води 0,5
см3 нітратної кислоти і 1 см3 розчину азотнокислого срібла
або до нейтральної реакції по метилоранжу (для промивання рекомендується
використовувати розчини і воду, нагріті до температури 60…80
°С);
- після
обробки катіоніт
КУ-2-8 в Н-формі зберігають під шаром дистильованої води в добре закритому
посуді не більше року.
Підготовка
річкового або морського піску
Пісок просівають, збирають фракцію 0,25…1,0 мм, відмочують водою і
промивають розчином хлоридної кислоти (1:3) до негативної реакції на іон заліза
з розчином роданіду амонію або роданіду калію. Потім пісок промивають
дистильованою водою для видалення іонів хлору і проводять перевірочну реакцію з
розчином нітратного срібла або з метилоранжем. Для
промивання рекомендується використовувати розчин кислоти і воду, нагріті до
температури 60…80 °С.
Очищений
пісок сушать і прожарюють при температурі 500…600 ° С протягом 5
годин.
Приготування
спиртово-кислотних сумішей
Готують
два види спиртово-кислотних сумішей.
Суміш
для осадження пектину:
до 100 см3
етилового спирту додають 2 см3
концентрованої хлоридної кислоти.
Суміш для промивання осаду пектину: 100 см3 70% - вого розчину етилового спирту змішують з 5 см3 концентрованої хлоридної кислоти.
Хід аналізу:
- беруть
наважку
в кількості 30…50
г для натуральних продуктів і 10…20
г для продуктів, приготованих з додаванням пектину (беруть
по дві наважки для визначення окремо пектину і протопектину);
- продукти, які містять доданий жир, попередньо знежирюють наступним
чином: наважку досліджуваного матеріалу поміщають в патрон з фільтрувального
паперу і підсушують при температурі 70…80 °С; висушену пробу поміщають в колбу
місткістю 250 або 500 см3 зі шліфом, заливають 30…40 см3
етеру і нагрівають на водяній бані при температурі 40…50 ° С зі
зворотним холодильником 20…30 хв.; етер обережно зливають або фільтрують через
паперовий фільтр, а відділення жиру повторюють ще чотири-п'ять разів (знежирення
проби можна проводити в апараті Сокслета); залишки проби з фільтром додають в
колбу з знежиреної наважкою, заливають 100 см3 підігрітою до
60…70 ° С дистильованою водою і далі проводять екстрагування
пектинових речовин;
1.Вилучення
водорозчинного пектину
- наважку
досліджуваного продукту поміщають в колбу місткістю 250 або 300
см3;
- заливають 100 см3 підігрітою до 60…70 ° С
дистильованою водою;
- струшують 30 хв.;
- потім вміст кількісно дистильованою водою переносять в мірну колбу
місткістю 200 або 250 см3;
- охолоджують, доводять до мітки, ретельно перемішують і відокремлюють
рідину центрифугуванням;
- отриманий екстракт водорозчинного пектину переносять в сухий
посуд;
- при
аналізі фруктових соків без м'якоті операцію вилучення водорозчинного пектину
поєднують з очищенням екстракту як описано нижче (п.3);
- для консервів з низькоетерифікованим пектином, при виробництві яких
використовується сіль кальцію, яка заважає кількісному виділеню пектинових
речовин, екстракцію водорозчинного пектину проводять у присутності 0,5…0,7 г
(1,5 см3) обробленого катіоніта КУ-2-8, який додається в колбу з
пробою;
2.Сумарний
вміст пектинових речовин
визначають
в інший наважці
продукту після проведення гідролізу у присутності хлоридної кислоти, для
переведення
протопектину у
розчинний стан. Для цього:
- наважку досліджуваного матеріалу поміщають в колбу місткістю 250 або 300
см3,
- заливають 100 см3 розчину хлоридної кислоти концентрації 0,05
моль/дм3 (рН суміші 1,8-2,0);
- нагрівають на водяній бані 30 хв. при температурі
85…90 ° С;
- потім вміст колби кількісно дистильованою водою переносять в мірну колбу
місткістю 200
або 250 см3;
- охолоджують, доводять до мітки, перемішують і залишають на 1…1,5 годин
для вирівнювання концентрації пектинових речовин в рідкій і твердій
фазах;
- екстракт відокремлюють центрифугуванням і збирають в сухий посуд;
3.Отримані розчини пектинових речовин очищають осадженням
спиртово-кислотної сумішшю. Для цього:
- в хімічний стакан з допомогою піпетки поміщають 25, 50 або 100
см3 екстракту (в залежності від вмісту
пектину);
- додають подвійну кількість спиртово-кислотної
суміші;
- ретельно перемішують і залишають на 1…1,5 години для формування осаду;
- при аналізі фруктових соків без м'якоті до наважки соку додають
двократний об'єм спиртово-кислотної суміші, ретельно перемішують і залишають на
1…1,5 години для формування осаду;
- осад, який випав
фільтрують через лійку з пористою
платівкою ВФ-1-40 ПОР 40 з шаром піску 0,5-0,7 см;
- склянку
і осад промивають розчином 70 % - вого
етилового спирту, підкисленого хлоридною кислотою три рази по 15…20
см3;
- потім
розчином 70 % - вого
етилового спирту до негативної реакції на іон хлору з нітратним сріблом;
- на
промивку однієї проби витрачається 90…100 см3
70 % - ного
розчину етилового спирту;
- лійку
з промитим осадом встановлюють в чисту колбу з тубусом місткістю 250
см3
і кількісно розчиняють пектиновмісний
осад водою за
температури
60…70 ° С;
- склянку,
в якій проводили
осадження, також промивають два-три рази підігрітою водою;
- охолоджують
розчин до кімнатної температури;
- додають
6 крапель індикатора Хінтона
і титрують розчином гідроксиду натру
концентрацією
0,05 моль/дм3
до переходу жовтого забарвлення в малинове,
яке
не зникає протягом 20…30 с.
- до
розчину в колбі за допомогою піпетки або бюретки додають 20 см3
розчину гідроксиду натру
концентрацією
0,1 моль/дм3;
- закривають
пробкою і залишають на 30 хв.;
- за допомогою бюретки доливають розчин хлоридної
кислоти концентрацією 0,1 моль/дм3, точну кількість якого
встановлюють попереднім титруванням 20 см3 гідроксиду натру
концентрацією 0,1
моль/дм3
тим же розчином кислоти з індикатором Хінтона;
- суміш у колбі знову титрують розчином гідроксиду натру концентрацією
0,05 моль/дм3;
- результат першого титрування пропорційний вмісту вільних, а другого -
етерифікованих карбоксильних груп і при множенні на відповідні еквіваленти
знаходять масову частку поліуронідної частини пектинових
речовин.
Розрахунки:
Масову частку поліуронідів (X) у відсотках обчислюють за формулою:
де,
V1,
V2
- об'єми розчину гідроксиду натру,
витрачені на перше і друге титрування, см3;
c -
точна концентрація розчину гідроксиду натру,
який використовується для титрування, моль/дм3 (0,05 моль/дм3,
помножена на поправочний коефіцієнт);
V -
загальний об'єм
екстракту, см3;
V3 -
об'єм
екстракту, відібраний для осадження і титрування, см3;
m -
маса наважки, г;
m1 -
молекулярна маса ланки полігалактуронової кислоти, m1 = 176 г/моль;
m2 -
молекулярна маса етерифікованої
ланки полігалактуронової кислоти, m2
=
190 г/моль;
Ступінь
етерифікації виділених пектинових речовин (ε) у відсотках обчислюють за
формулою:

Кількість
водонерозчинного пектину (протопектину) визначають за різницею між загальним
вмістом пектинових речовин (п.2)
і вмістом
водорозчинного пектину (п.1).
За
кінцевий результат випробувань приймають середнє
арифметичне
результатів двох паралельних визначень, допустима
абсолютна розбіжність між якими не повинна
перевищувати 0,10% при визначенні поліуронидів
і 4,0% при визначенні ступеня етерифікації (Р =
0,95).
9.3.3 Визначення вмісту
пектинових речовин карбазольним методом
Метод поширюється на фруктові і овочеві соки, нектари, соковмісні напої,
фруктові та овочеві концентровані соки, пюре та концентровані пюре, морси і
концентровані морси, сокову продукцію з фруктів і овочів збагачену і для
дитячого харчування (далі – сокова продукція) і встановлює
фотометричний метод визначення масової частки загального пектину і
водорозчинного пектину.
Діапазони визначення загального пектину і водорозчинного пектину в сокової продукції - від 70 до 3500 мг / дм3 або масової частки від 250 до 3500 млн-1 включно.
Суть методу
Полягає у фотометричному визначенні пектину при довжині хвилі 525 нм.
Масову концентрацію пектину в сокової продукції визначають на основі реакції
карбазолу з окремими фракціями пектину в присутності концентрованої сульфатної
кислоти, з подальшим вимірюванням поглинання світла.
Осадження всіх фракцій пектину здійснюють шляхом обробки проби етиловим
спиртом. Одержуваний сумарний осад може бути використаний для визначення
загального пектину або, після відповідних обробок, для селективного визначення
окремих фракцій. Для кількісного визначення використовують реакцію карбазолу з
окремими фракціями пектину, які містяться в екстрактах, в присутності
концентрованої сульфатної кислоти.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: технічні терези, спектрофотометр або ФЕК з фільтром для вимірювання
екстинкції за довжини хвилі 525…530 нм, термометр з діапазоном вимірювання від 0
°С до 150 °С з похибкою ± 1 °С, водяна баня, бюретки, піпетки, мірні колби на 50
та 100 см3, конічні колби на 150 см3, пробірки, штатив,
секундомір, пробірки центрифужні градуйовані, місткістю 50 см3
конічним дном, центрифуга, шейкер горизонтальний, ексикатор, папір фільтрувальний, палички
склянні.
Реактиви: вода дистильована, спирт етиловий ректифікований (95%); кислота сульфатна; фосфору оксид;
розчин 250 мг Na2B4O7•10H2O в 100 см3 H2SO4 густиною 1,84: хімічно чисту сульфатну кислоту прогрівають до початку виділення
SO2, після чого 0,15 см3 хімічно чистої
сечовини;
спирт етиловий ректифікований з харчової сировини
(63%): 200 см3 етилового ректифікованого спирту розбавити в 100
см3 дистильованої води, термін зберігання в щільно закупореній
упаковці - не більше 3 міс;
амоній щавлевокислий 1-водний
(0,75%): 7,5 г амонію щавлевокислого 1-водного перенести кількісно в мірну
колбу місткістю 1 дм3 і довести до мітки дистильованою водою. Термін
зберігання - не більше 1 міс.;
їдкий натр (1 моль/дм3): 4 г розчинити в 50 см3 дистильованої води і довести до 100
см3 дистильованою водою. Зберігати до помутніння, не більше одного
року;
карбазол, спиртовий розчин з масовою часткою
0,1%: 0,1 г сублімованого карбазолу розчиняють в етиловому спирті (95 %) в
мірній колбі місткістю 100 см3 і доводять до мітки, зберігають - не
більше 3 міс;
суміш, що складається з 1 см3
дистильованої води, 0,5 см3 спиртового розчину карбазолу (масової
концентрації 0,1%) і 6 см3 концентрованої сульфатної кислоти, повинна
бути прозорою;
Моногідрат галактуронової кислоти (не менше
97%): висушують під вакуумом не менше 5 годин при 30 °С або при
20 °С над пентокисом фосфору;
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Хід аналізу:
1.Звільнення наважки від цукрів, екстракція водорозчинного
пектину
- наважку масою 0,5 г (тонкі зрізи з різних частин плоду) переносять у
конічну колбу на 150 см3;
- заливають 25 см3 спирту ( для інтенсивно забарвлених плодів
беруть 70 см3 спирту, а для слабозабарвлених – 50
см3);
- кип'ятять зі зворотним повітряним холодильником 30…40
хвилин;
- осад відфільтровують і ще раз заливають 15 см3 спитру та
ставлять на 15 хв. на баню;
- відфільтровують, осад знову заливають 10 см3, екстрагують 10
хв., відфільтровують;
- у темнозабарвлених плодах операцію екстрагування та фільтрування
виконують 5 разів;
- фільтр підсушують;
- осад без втрат переносять у конічну колбу додаючи 40 см3
дистильованої води;
- колбу нагрівають на водяній бані при температурі 50 °С протягом 30 хв.
екстрагуючи водорозчинний пектин;
- осад відфільтровують у мірну колбу на 50 см3, змивають фільтр
невеликими порціями дистильованої води, фільтрують, доводять мітки, охолоджують
колбу до 20 °С;
- в отриманій витяжці визначають водорозчинний
пектин.
2.Вилучення (екстрагування) протопектину
- осад із фільтра переносять в екстракційну колбу;
- заливають 30 см3 1 н сульфатної кислоти і нагрівають на
водяній бані протягом 1 години;
- розчин охолоджують, фільтрують у мірну колбу на 100 см3,
доводять до мітки сульфатною кислотою;
- в отриманій витяжці визначають масову частку протопектину.
3.Проведення реакції з карбазолом
- у три пробірки ( 1 пробірка контрольна, в яку додають всі реактиви,
дистильовану воду, крім витяжки) вносять по 0,5 см3 досліджуваного
розчину (пектину чи протопектину);
- пробірки ставлять у посуд із льодом;
- обережно, по краплям по стінці пробірки додають 3 см3
концентрованої сульфатної кислоти з боратом не допускаючи нагрівання
суміші;
- пробірки необхідно постійно струшувати у воді з льодом, щоб не допустити
нагрівання;
- пробірки ставлять у киплячу водяну баню на 6 хв.;
- потім швидко охолоджують водою з льодом;
- в кожну пробірку додають по 0,1 см3 0,2 %-вого розчину
карбозолу;
- пробірки ставлять на киплячу водяну баню на 10
хв.;
- суміш охолоджують і проводять вимірювання оптичної густини розчинів на
ФЕК, товщиною шару розчину 5 мм при довжині хвилі 535 мкм, використовуючи
зелений світофільтр;
- аналогічно готують одну пробірку з розчинником для встановлення приладу
на нуль;
- будують калібрувальний графік.
Побудова калібрувального графіку
- калібрувальну криву будують за галактуроновою
кислотою;
- для отримання точних результатів конфентрація галактуронової кислоти в
екстракті повинна бути від 4 до 50
мкг в 0,5 см3;
- у цих межах інтенсивність забарвлення прямо пропорційна концентрації
галактуронової кислоти (табл. 9.4)
Таблиця 9.4
Оптична густина розчину, яка відповідає вмісту галактуронової
кислоти
|
Галактуронова
кислота, мкг |
Оптична
густина |
Галактуронова
кислота, мкг |
Оптична
густина |
|
4 |
0,055 |
25 |
0,238 |
|
6 |
0,073 |
30 |
0,282 |
|
8 |
0,091 |
35 |
0,325 |
|
10 |
0,102 |
40 |
0,368 |
|
15 |
0,152 |
45 |
0,410 |
|
20 |
0,195 |
50 |
0,454 |
Розрахунки:
Масову частку водорозчинного пектину чи протопектину розраховують за формулою:
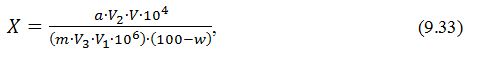
де X
– масова частка водорозчинного пектину чи протопектину в перерахунку на
суху речовину, %,
a
– вміст галактуронової кислоти, визначений за калібрувальною кривою, мкг,
V
– об'єм екстракту, отриманий із наважки, см3,
V2
– об'єм,
отриманий після розведення, см3,
m –
маса наважки, г
V3
– об'єм проби, взятої для реакції з карбазолом,
см3,
V1
– об'єм, взятий для розведення, см3,
104
– коефіцієнт переведення у відсотки,
106
– коефіцієнт переведення у грами,
100
– коефіцієнт перерахунку на 100 г сухої речовини,
w
– масова частка вологи, %.
За кінцевий результат випробувань приймають середнє арифметичне результатів двох паралельних визначень, допустима абсолютна розбіжність між якими не повинна перевищувати 0,10% при визначенні поліуронидів і 4,0% при визначенні ступеня етерифікації (Р = 0,95).
9.3.4 Особливості
визначення пектину в соковій продукції
1.Осадження пектину:
- 15 см3 сокової продукції переносять в центрифужну пробірку
або 4 г концентрованого соку або пюре переносять в центрифужную пробірку і
додають 11 см3 дистильованої води (об'єм проби може змінюватися в
залежності від передбачуваного вмісту пектину);
- об'єм проби в пробірці доводять до 40 см3 за допомогою
гарячого (75 °С) етилового спирту (95%), вміст перемішують скляною паличкою;
- паличку споліскують етиловим спиртом (95 %) у центрифужную пробірку і
доводять загальний об'єм до 50 см3 цим же розчином етилового
спирту;
- після температурної обробки пробірку з пробою центрифугують протягом 15
хв. при 5000 об хв.
- після центрифугування пробу декантують, відкидаючи надосадову рідину
(супернатант);
- до отриманого осаду додають до 40 см3 розчину етилового
спирту (63 %), вміст перемішують скляною паличкою;
- центрифужну пробірку з пробою нагрівають на водяній бані при температурі
85 °С протягом 10 хв., ретельно перемішуючи вміст скляною паличкою, виключаючи
закипання розчину;
- паличку споліскують розчином етилового спирту (69 %) в центрифужную
пробірку і доводять об'єм до 50 см3 цим же
розчином;
- після температурної обробки (85 °С) пробірку поміщають в центрифугу, в
якій центрифугируют пробу протягом 15 хв. при 5000 об /
хв.
- після центрифугування пробу декантують, відкидаючи супернатант.
2.Екстракція загального
пектину
- для
визначення загального пектину, отриманий осад, кількісно переносять у
мірну колбу місткістю 100 см3,
- додають
5 см3
розчину гідроксиду натру
молярної концентрації 1 моль/дм3;
- доводять
до мітки дистильованою водою;
- вміст перемішують і залишають на 15 хв. при кімнатній температурі,
періодично струшуючи колбу;
- розчин фільтрують через фільтр;
- отриманий екстракт використовують для визначення концентрації загального пектину в пробі при проведенні вимірювань фотометричним методом.
3.Екстракція водорозчинного пектину:
- 5 см3 дистильованої води додають до осаду у центрифужній
пробірці, отриманого по п. 1.
- додають в центрифужну пробірку половину чайної ложки подрібненого і
зволоженого фільтрувального паперу;
- вміст ретельно перемішують за допомогою скляної
палички;
- частинки осаду, що залишилися на паличці, змивають у центрифужну
пробірку дистильованою водою;
- об'єм вмісту пробірки доводять до 35 см3 дистильованою
водою;
- вміст пробірки інтенсивно перемішують за допомогою шейкера (~
200…300 хв-1) протягом 10 хв.
- пробірку поміщають в центрифугу, в якій проводять центрифугування
протягом 15 хв. при 5000 об/хв.
- після центрифугування надосадову рідину декантують через складчастий
фільтр в мірну колбу місткістю 100 см3 або 200
см3;
- 10 см3 дистильованої води додають до осаду в центрифужній
пробірці,
- вміст ретельно перемішують за допомогою скляної
палички;
- частинки осаду, що залишилися на паличці, змивають у центрифужну
пробірку дистильованою водою;
- об'єм вмісту пробірки доводять до 20 см3 дистильованою
водою;
- вміст пробірки інтенсивно перемішують за допомогою шейкера
(~ 200…300 хв-1) протягом 10 хв.;
- пробірку поміщають в центрифугу, в якій проводять центрифугування
протягом 15 хв. при 5000 об/хв.
- після центрифугування надосадову рідину декантують через складчастий
фільтр в ту ж мірну колбу місткістю 100 см3 або 200
см3;
- екстрагування дистильованою водою повторюють двічі;
- у мірну колбу із зібраним екстрактом додають 5 см3 або 10 см3 розчину гідроксиду натру молярної концентрації 1 моль/дм3, об'єм вмісту колби доводять дистильованою водою до мітки;
*Примітка: вибір мірної колби залежить від вмісту водорозчинного пектину в аналізованої пробі (наприклад, якщо вміст водорозчинного пектину в сокової продукції до 450 мг/дм3 (або до 1800 мг/дм3 в концентрованій пробі), то використовують мірну колбу місткістю 100 см3, якщо вміст 450…900 мг/дм3 (або 1800…3500 мг/дм3 в концентрованій пробі) - мірна колба місткістю 200 см3. Відповідно змінюється кількість доданого розчину гідроксиду натру молярної концентрації 1 моль/дм3.
- отриманий екстракт використовують для визначення масової концентрації водорозчинного пектину в пробі при проведенні вимірювань фотометричним методом.
4.Проведення вимірювань фотометричним
методом
- у дві лабораторні пробірки "проба" і "контроль" піпеткою вносять по
1 см3 екстракту, отриманого згідно з п.2;
- додають в пробірку "проба" 0,5 см3 спиртового розчину
карбазолу масової концентрації 0,1%.
- в пробірку "контроль" вносять 0,5 см3 етилового спирту (95
%);
- у пробірках з пробою формується осад, у вигляді білих
пластівців;
- в обидві пробірки ("проба" і "контроль") додають по 6 см3 концентрованої сульфатної кислоти при постійному перемішуванні скляною паличкою довжиною 200 мм;
* Примітка: 6 см3 сульфатної кислоти необхідно рівномірно влити за 7 с. Ця умова обов'язкова, так як саме за цей проміжок часу розчин в пробірці досягає температури 85 °С, необхідної для отримання його стабільного забарвлення, і уникає інтенсивного закипання.
- пробірки негайно поміщають у водяну баню з температурою 85 °С на 5 хв.
при постійному помішуванні;
- охолоджують у водяній бані температурою 20 °С протягом 15
хв.;
- протягом не більше 20 хв. вимірюють оптичну густину розчину проби в пластикових одноразових кюветах на спектрофотометрі при довжині хвилі 525 нм по відношенню до "контролю".
**Для виключення можливих помилок, пов'язаних з реакцією розчину
карбазолу, проводять контрольне визначення тільки з даними розчином (з
дистильованою водою замість проби). Для цього готують чотири розчини, оптичну
густину яких вимірюють один проти одного:
a)
1 см3 екстракту + 0,5 см3 спиртового розчину
карбазолу + 6 см3 сульфатної кислоти;
b)
1 см3 екстракту + 0,5 см3 етилового спирту + 6
см3 сульфатної кислоти;
c)
1 см3 дистильованої води + 0,5 см3 спиртового
розчину карбазолу + 6 см3 сульфатної кислоти;
d)
1 см3 дистильованої води + 0,5 см3 етилового
спирту + 6 см3 сульфатної кислоти.
Оптичну густину розчину "а" вимірюють проти розчину "b" (значення "x"). Оптичну густину розчину "c" вимірюють проти розчину "d" (значення "y"). Значення масової концентрації (масової частки) пектину (виражене через масовому частку ангідриду галактуронової кислоти) ("x-y") визначають за калібрувальним графіком.
5.Побудова калібрувального графіку
5.1 Приготування основного калібрувального розчину
- 120,5 мг моногідрату галактуроновой кислоти, кількісно переносять в
мірну колбу місткістю 1 дм3;
- у колбу додають 0,5 см3 розчину гідроксиду натру молярної
концентрації 1 моль/дм3,
- потім доводять об'єм до позначки дистильованою
водою;
- після перемішування вмісту колбу залишають на ніч при кімнатній
температурі;
- отриманий основний калібрувальний розчин містить 100 мг ангідриду
галактуроновой кислоти (ДКА) в 1 дм3;
- термін зберігання розчину - не більше 3 міс в темному місці при кімнатній температурі.
5.2Приготування робочих калібрувальних
розчинів
- готують серію калібрувальних розчинів, масова концентрація ангідриду
галактуроновой кислоти в яких становить 10, 30, 40, 50 і 70 мг/дм3,
- раствори готують в мірних колбах місткістю 100 см3 в які
вносять відповідно 10, 30, 40, 50 і 70 см3 основного калібрувального
розчину;
- доводять об'єм розчину до мітки дистильованою
водою;
- розчини використовуються свіжоприготовленими.
Побудова калібрувальної
характеристики
- виконують операції відповідно до п 4, вводячи замість проби по 1
см3 калібровочних розчинів, приготованих за п. 5.1 та
5.2;
- вимірюють оптичну густину розчинів: у кожній точці проводять не менше
трьох вимірів;
- за результат вимірювання оптичної густини кожного калібрувального
розчину приймають середньоарифметичне значення трьох
вимірів.
- калібрувальну характеристику будують один раз на рік, а також при зміні реактивів або зміні умов аналізу.
Розрахунок
Розрахунок масової концентрації пектину С, мг/дм3, обчислюють за
формулою:

де Ck – масова концентрація ангідриду галактуронової кислоти, яка визначається
за калібрувальним графіком, мг/дм3;
V2 - об'єм розчину, приготованого для фотометричного визначення,
см3;
V1- об'єм сокової продукції, взятої для аналізу,
см3.
Розрахунок масової частки пектину С в концентрованому соці або пюре,
млн-1, обчислюють за формулою:

де Ck – концентрація ангідриду галактуронової кислоти, яка визначається за
калібрувальним графіком, мг/дм3;
V2 - об'єм розчину, що приготований для фотометричного визначення,
см3;
m - маса концентрованого соку або пюре, взятого для аналізу,
г;
Масова концентрація пектину виражається в мг ГКА в 1 дм3
(мг/дм3), масова частка - в мг ГКА в 1 кг
(млн-1).

де M2
–
маса висушеного фільтрату з осадом пектату кальцію,
г,
M1
–
маса висушеного фільтра, г,
0,9235
– коефіцієнт перерахунку на пектинову кислоту;
V
– об'єм фільтрату, см3,
V1
– об'єм розчину, взятого для визначення, см3,
m
– маса наважки, г.
Розрахунки виконують з точністю до 0,01 %. Маса осаду не повинна перевищувати 0,03 г. Якщо більше, то осад важко висушується, а результати отримують завишені (розчин 2 аналізують подібним чином).