8.ВИЗНАЧЕННЯ КИСЛОТНОСТІ ПЛОДІВ І ОВОЧІВ ТА ПРОДУКТІВ ЇХ ПЕРЕРОБКИ
Кислотність
– важливий показник якості як свіжої, так і переробленої плодоовочевої і ягідної
продукції. Вона має вирішальну роль при оціюванні смакових властивостей
продукції. У готових продуктах визначенню кислотності належить вагоме місце,
оскільки вона не тільки визначає смакові якості, але й є показником його
свіжості та доброякісності.
Активна
кислотність (рН)
показує ступінь дисоціації кислот. Активна кислотність має важливе технологічне
значення. Вона характеризує ступінь вираженості смаку, за нею визначають рівень
температури стерилізації кислотних або слабкокислотних
продуктів.
Титрована
кислотність –
це кількість вільних органічних кислот і їх кислих солей, які знаходяться у
досліджуваному продукті. До основних органічних кислот відносять оцтову,
молочну, яблучну, лимонну, щавлеву, винну, тощо.
8.1 Визначення активної кислотності (рН) потенціометричним методом
Метод
поширюється на свіжі овочі, фрукти та ягоди, продукти їх переробки, в тому числі
на сокову продукцію, м'ясні та м'ясо-рослинні консерви (далі–товари) і дозволяє
визначити величини рН в діапазоні вимірювань від 2 до 12 од. рН
включно.
Суть методу
Полягає у вимірюванні різниці потенціалів між двома електродами
(вимірювальним і електродом порівняння), зануреними в досліджувану пробу.
Виміряне значення виражають в одиницях рН (од. рН).
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали: рН-метр або рН-метр-іономер зі скляним і хлорсрібним електродами або комбінованим електродом з діапазоном вимірювань активності іонів водню від 0 до 14 од. рН і межею абсолютної похибки вимірювань ± 0,05 од. рН, що оснащений системою (функцією) термокомпенсації (рис. 28);

терези неавтоматичної дії спеціального класу точності з
максимальним навантаженням 200 або 220 г і ціною повірочної поділки ± 0,001 г;
колба мірна; склянки на 50 та 100 см3; лійки скляні і лійки ділильні;
папір фільтрувальний лабораторний; вата медична гігроскопічна; термометр
рідинний скляний з межею допустимої похибки ± 1,0 ° С в діапазоні вимірювань від 0 ° С до 55 ° С; етер діетиловий медичний: перед застосуванням етер насичують
дистильованою водою (в об'ємному співвідношенні етеру і води – 8:1) шляхом
струшування і подальшого відділення верхнього шару; спирт етиловий
ректифікований, розведений дистильованою водою в об'ємному співвідношенні
етанолу і води – 3:2; стандарт-титри для приготування буферних розчинів 2-го
розряду з номінальними значеннями 4,01; 6,86 і 9,18 од. рН при температурі 25 ° С; Вода дистильована, або деіонізована, або вода для лабораторного
аналізу будь-якого ступеня чистоти зі значенням рН від 5,0 до 7,5
од. рН.
Реактиви
для приготування буферних розчинів
(див. додаток Б, таблицю Б 1):
-
калій виннокислий кислий з масовою часткою основної речовини не менше
99%;
-
калій фталевокіслий кислий з масовою часткою основної речовини не менше 99%;
-
калій фосфорнокислий однозаміщений, х.ч. або ч.д.а.;
-
натрій (бура) 10-водний тетраборнокислий;
-
натрій фосфорнокислий двозаміщений 12-водний, х.ч. або
ч.д.а.
Допускається
приготування буферних розчинів з інших солей.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні.
Підготовка приладу:
Готують прилад до роботи і проводять його градуювання і (або) контроль
показань відповідно з керівництвом (інструкцією) по експлуатації, використовуючи
буферні розчини, приготовлені з стандарт-титрів або реактивів. При відсутності
стандарт-титрів буферних розчинів використовують буферні розчини, наведені в
додатку Б, таблиці Б 1.
Підготовка електродів:
Перед вимірами електроди ретельно промивають дистильованою водою і
висушують (НЕ протираючи) залишки води фільтрувальної
папером.
Після вимірювань проб, які містять жир, електроди очищають ватяним
тампоном, змоченим діетиловим етером, насиченим дистильованою водою або етиловим
спиртом, а потім дистильованою водою. Допускається використання готових розчинів
для очищення електродів відповідно до рекомендацій виробника.
Зберігання електродів здійснюють відповідно до рекомендацій
виробника.
Хід аналізу:
Проводять два паралельних вимірювання в умовах
повторюваності.
1. 20г підготовленого середнього зразка переносять, змиваючи гарячою
дистильованою водою (80 °С) в мірну колбу місткістю 250 см3 та
залишають на 30 хвилин періодично збовтуючи. Потім охолоджують, доливають
домітки і добре перемішують. Рідину фільтрують через сухий складчастий фільтр
або вату в суху склянку.
2. У склянку поміщають лабораторну пробу (фільтрат) в кількості, достатній
для занурення електродів.
3. Тверді лабораторні проби і лабораторні проби густої консистенції
попередньо розбавляють приблизно в два рази дистильованою водою. У консервах, що
мають тверду і рідку фазу, допускається проводити вимірювання безпосередньо в
рідкій фазі продукту.
4. При використанні приладу, незабезпеченого системою термокомпенсации,
температура повинна бути (20 ± 5) ° С.
5. У склянку з пробою занурюють стрижень магнітної мішалки і встановлюють
стакан на магнітну мішалку (див. рис. 28). Вмикають двигун мішалки.
6. Занурюють електроди на глибину не менше 30 мм і регулюють швидкість
обертання стрижня таким чином, щоб виключити утворення лійки. При цьому
електроди не повинні торкатися стінок і дна склянки.
7. Перемішування проби слід проводити нетривалий час (як правило, протягом
30-60 с).
8. Після перемішування вмісту склянки магнітну мішалку вимикають і дають
пробі стабілізуватися.
9. Потім вмикають рН-метр. Після того як значення рН встановиться,
реєструють його за шкалою або за цифровим індикатором приладу. Зняття показань
слід проводити не пізніше ніж через 5 хв після занурення
електродів.
10. Після вимірювань кожної проби електроди промивають кілька разів водою,
залишки якої видаляють обережним висушуванням фільтрувальним
папером.
Розрахунки:
Вимірювання повторюють 2…3 рази, кожного разу виймаючи електроди з розчину і при вимірюванні знову їх занурюючи. Різниця між паралельними визначеннями не повинна перевищувати 0,1 одиниці рН. Остаточне значення рН приймається як середнє арифметичне всіх значень.
8.2 Визначення загальної (титрованої)
кислотності
Два
описаних нижче методи – метод титрування в присутності кольорового індикатора та
потенціометричний референтний метод – дозволяють визначити титровану
кислотність у свіжих фруктах, овочах та ягодах, а також продуктах їх переробки.
Метод
титрування в присутності кольорового індикатора не застосовують при аналізі вин.
При
аналізі деяких забарвлених продуктів важко визначити кінцеву точку титрування і
тоді застосовується потенціометричний
референтний
метод.
При
аналізі продуктів з додаванням двооксиду сірки титрована
кислотність не може бути правильно оцінена через присутність двооксиду
сірки.
8.2.1 Титрометричний метод визначення загальної кислотності
(арбітражний)
Суть методу
Полягає у нейтралізації органічних кислот, які знаходяться у дослідному
продукті, 0,1 н розчином лугу. Титрування виконують до переходу розчину з
кислого середовища в лужне. Момент переходу середовища в лужне візуально
фіксується за появою рожевого забарвлення розчину в присутності индикатора
фенолфталеїну. Точність методу ±0,5%.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали.
Гомогенізатор, блендер або ступка з товкачиком лабораторні; піпетки
місткістю 25, 50 або 100 см3; колба конічна, до якої можливе
приєднання зворотного холодильника; колба мірна місткістю 250
см3;стакан місткістю 250 см3 з магнітною або механічною
мішалкою; бюретка місткістю 50
см3; холодильник зворотний; терези аналітичні з точністю зважування
до 0,01 г; баня водяна.
Реактиви:
для
проведення аналізу використовують реактиви лише встановленої аналітичної чистоти
і дистильовану або демінералізовану воду, або воду еквівалентної чистоти: натру
гідроокис NaOH концентрацією 0,1 моль/дм3
(0,1 н); фенолфталеїн, розчин масовою концентрацією 10
г / дм3(1%-вий) в етиловому спирті об'ємною концентрацією
95%.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні.
Підготовка
проб для аналізу:
1.Рідкі
продукти (продукти, які містять тільки рідку фазу, наприклад,
соки, консервовані фруктові сиропи, маринадні заливки, розсоли, рідина з
ферментованих продуктів):
- частину
попередньо перемішаної лабораторної проби фільтрують через вату, паперовий
фільтр або тканину;
-
в
мірну колбу на 250 см3 піпеткою вносять 25 см3 фільтрату;
-
доводять
водою до мітки та старанно струшують;
-
можна
відібрати пробу для аналізу за масою, зважуючи з точністю до 0,01 г не
менше 25 г лабораторної проби.
З проби газованих рідких продуктів перед аналізом
видаляють вуглекислий газ струшуванням проби протягом 3...4 хв при зниженому
тиску.
2.Продукти, крім рідких
харчових продуктів:
-
з проби видаляють плодоніжки, кісточки,
щільні стінки насіннєвих камер і, де можливо, зернятка (при аналізі заморожених
або глибокозаморожених продуктів це роблять після розморожування),
ретельно перемішують;
Розморожування заморожених або
глибокозаморожених продуктів проводять в закритих судинах, рідину, яка
утворюється при цьому додають до продукту перед змішуванням або
подрібненням.
При аналізі сушених продуктів частину
лабораторної проби ріжуть на дрібні шматочки.
- далі
проби гомогенізують або подрібнюють в ступці;
- зважують
з точністю до 0,01 г не менше 25 г лабораторної проби і переносять її в конічну
колбу та додають 50 см3 гарячої води;
- ретельно
перемішують до отримання однорідної консистенції;
- приєднують
до конічної
колби зворотний холодильник і нагрівають колбу з вмістом на водяній бані
за температури 80° С 15...30
хв (якщо продукт гомогенізований, то теплову обробку можна обмежити 5
хвилинами);
- вміст
колби охолоджують і кількісно переносять в мірну колбу на 250 см3 і
доводять водою до мітки, ретельно перемішують і
фільтрують.
Якщо
потрібно перевірити виконання вимог повторюваності, то середні проби готують
двічі.
Хід аналізу:
1. У склянку з мішалкою вносять піпеткою підготовлену пробу для аналізу
об'ємом 25, 50 або 100 см3 в залежності від очікуваної кислотності;
2. Додають у склянку 0,25…0,5 см3 розчину фенолфталеїну;
3. За постійного перемішування, виконують титрування з бюретки розчином
гідроксиду натру до появи рожевого забарвлення, яке не зникає протягом 30
с;
4. Фіксують кількість лугу, яка витрачена на титрування.
Розрахунки:
1.Обробка
результатів визначення для проби, що
взята за об'ємом (рідкі продукти):
Титровану кислотність Т (у 100г на 100
см3, або ммоль Н+ на 100 см3, або у %) з
урахуванням розведення, обчислюють за формулою:
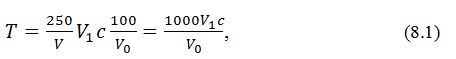
де
V-
об'єм аналізованої проби, наприклад 25 см3, см3;
V1
– об'єм гідроксиду натру, який витрачено на титрування, см3;
V0
– об'єм
проби для аналізу, см3;
c – точна
концентрація гідроксиду натру, моль/дм3;
250 – об'єм мірної колби,
см3;
.....100
– коефіцієнт для розрахунку титрованої кислотності на 100 г
продукту;
1000 – коефіцієнт,
отриманий при розрахунку:
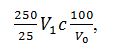
Обчислення
виконують до другого десяткового знака. Результат округлюють до першого
десяткового знака.
2.Обробка
результатів аналізу для проби, що взята
за масою:
Титровану кислотність Т (у 100г на 100 см3, або ммоль Н+ на 100 см3, або у %), з урахуванням розведення, визначають за формулою:
де
m –
маса наважки, г;
V1
– об'єм гідроксиду натру, який витрачено на титрування, см3;
V0
– об'єм
проби для аналізу, см3;
c – точна
концентрація гідроксиду натру, моль/дм3;
250 – об'єм мірної
колби, см3;
.....100
– коефіцієнт для розрахунку титрованої кислотності на 100 г
продукту;
Обчислення
проводять до другого десяткового знака. Результат округлюють до першого
десяткового знака та заносять у лабораторний журнал (робочий зошит) (табл. 8.2).
3.Альтернативний
спосіб представлення результатів визначення:
За необхідності допускається альтернативний спосіб представлення результатів визначення титрованої кислотності у грамах відповідної кислоти на 100 г або 100 см3 шляхом множення результату, отриманого за формулами 8.1 або 8.2, на коефіцієнт для відповідної кислоти за таблицею 8.1.
Таблиця 8.1
Коефіцієнти перерахунку на відповідну кислоту
|
Найменування кислоти |
Коефіцієнт
перерахунку |
Найменування
продуктів, які перераховують на дану кислоту |
|
Яблучна |
0,067 |
більшість
фруктів, ягід та овочів свіжих і перероблених |
|
Щавлева
|
0,045 |
щавель,
ревінь, шпинат |
|
Винна
|
0,075 |
виноград |
|
Сульфатна |
0,049 |
сульфітовані
продукти |
|
Оцтова
|
0,060 |
маринади |
|
Молочна
|
0,090 |
солено-квашені
продукти |
|
Лимонна
|
0,064 |
цитрусові
плоди, гранатник, смородина |
Таблиця 8.2
Форма запису результатів у лабораторний журнал
|
№
зразку |
Маса
наважки, г, m |
Об'єм
проби для аналізу, см3, V0 |
Об'єм
гідроксиду натру, який витрачено на титрування, см3, V1 |
Титрована
кислотність, %, Т |
Титрована
кислотність у перерахунку на необхідну кислоту, %, Т1 |
|
|
|
|
|
|
|
8.2.2Потенціометричний референтний метод визначення
титрованої кислотності
Суть методу
Полягає у потенціометричному
титруванні аналізованого розчину розчином гідроксиду натру.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: гомогенізатор, блендер або ступка з товкачиком лабораторні; піпетки
місткістю 25,
50 або 100 см3; колба
конічна,
до якої можливе приєднання зворотного холодильника; колба
мірна
місткістю 250 см3;стакан місткістю 250 см3 з магнітною або
механічною мішалкою; бюретка
місткістю 50 см3; холодильник зворотний; терези аналітичні з точністю
зважування до 0,01 г; баня
водяна;
рН-метр з точністю вимірювання не менше 0,05 од. рН.
Реактиви:
для
проведення аналізу використовують реактиви лише встановленої аналітичної чистоти
і дистильовану або демінералізовану воду, або воду еквівалентної чистоти: натру
гідроокис NaOH концентрацією 0,1 моль/дм3
(0,1 н); фенолфталеїн, розчин масовою концентрацією 10
г / дм3(1%-вий) в етиловому спирті об'ємною концентрацією
95%, буферні розчини з відомим значенням рН.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні.
Підготовка проб для аналізу
Виконують
як описано у пепередній методиці (арбітражний метод).
У
склянку з мішалкою вносять піпеткою пробу для аналізу, підготовлену та
розбавлену як описано вище, об'ємом 25, 50 або 100 см3 в залежності
від очікуваної кислотності.
Перевірка
правильності роботи рН-метра: правильність
роботи рН-метра перевіряють, використовуючи буферні розчини.
Хід аналізу
1. У
розчин занурюють електроди рН-метра;
2. Вмикають
мішалку і перемішують
вміст склянки;
3. Не припиняючи перемішування, додають з бюретки розчин гідроокису натру спочатку швидко, поки значення рН, не досягне (7,0 ± 0,2) од. рН, а потім повільно, поки значення рН не досягне (8,1 ± 0,2) од. рН.
Розрахунки:
Загальну титровану кислотність виражають у відсотках (у 100 г на 100
см3) в перерахунку на відповідну кислоту за
формулою:

де
m –
маса наважки, г;
V1
– об'єм гідроксиду натру, який витрачено на титрування, см3;
V0
– об'єм
проби для аналізу, см3;
К – коефіцієнт
перерахунку на відповідну кислоту (табл. 8.1);
250 – об'єм мірної колби, см3;
...100 – коефіцієнт для розрахунку титрованої кислотності на 100 г продукту;
Обчислення
проводять до другого десяткового знака. Результат округлюють до першого
десяткового знака та заносять у лабораторний журнал (робочий зошит) (табл.
8.2).
8.2.3 Визначення титрованої кислотності сусла
(вин)
Суть методу
Полягає у прямому титруванні визначеного об'єму сусла титрованим
розчином лугу до нейтральної реакції, яка встановлюється за допомогою
індикатору.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали:
конічна колба на 250 см3, бюретка на 25 см3,
піпетка на 10 см3, скляна паличка, плитка електрична.
Реактиви: 0,1 н розчин їдкого натру, 1 н розчин їдкого натру, 0,4 %-вий розчин
бромтимолового синього ( 0,4 г індикатора розчиняють у 10 см3
спирту – ректифікату та доводять свіжокип'яченою, дистильованою водою
(рН=7) до 100 см3); буферний розчин з рН =7 ( 107,3 г однозаміщеного
фосфорнокислого калію розчиняють у 500 см3 1 н розчину гідроокису
натру та доводять водою до обєму 1000 см3).
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні.
Хід аналізу
- у конічну колбу відбирають 10 см3 сусла (або
вина);
- додають 25 см3 води;
- нагрівають до початку кипіння з метою видалення вуглекислого
газу;
- до проби додають 1 см3 індикатору бромтимолового
синього;
- тирують 0,1 н розчином їдкого натру до появи зелено-синього
забарвлення;
- далі без перерви додають 5 см3 буферного розчину,
рН=7;
- отриманий розчин є розчином для порівняння, який можна використовувати
для серії визначень кислотності сусел (вин) близьких за
кислотністю;
- в другу колбу відбирають 10 см3 сусла (або
вина);
- додають 30 см3 води;
- нагрівають до кипіння;
- до проби додають 1 см3 індикатору бромтимолового
синього;
- тирують 0,1 н розчином їдкого натру до появи забарвлення, що ідентичне забарвленню розчину для порівняння.
Розрахунки:
Титровану кислотність визначають у міліграм-еквівалентах (мг-екв) на літр, або в грамах на літр у перерахунку на винну, сульфатну або яблучну (для плодово-ягідних вин) кислоту за формулою:

де T
– тирована кислотність, мг-екв/л,
а
– кількість 0,1 н розчину їдкого натру витрачена на тирування,
см3,
v
– об'єм проби, см3,
1000
–
множник для перерахунку на 1000
см3,
Показник К показує кількість міліграмів – еквівалентів або грамів
кислоти, яка відповідає 1 см3 розчину їдкого натру. Для 1 см3 0,1 н розчину К дорівнює 0,1
мг–екв, або 0,0075 г виної, 0,0067 яблучної та 0,0049 г сульфатної кислот.
Підставляючи ці значення у формулу 8.4 та приймаючи що v=10 см3, отримуємо: для виної кислоти:
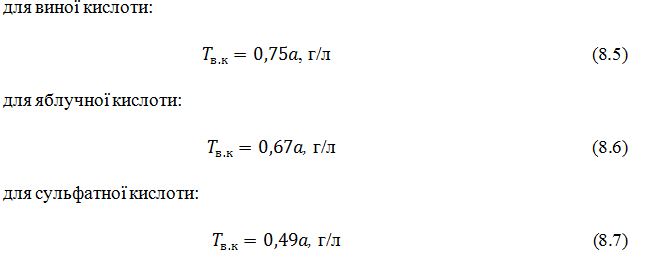 +
+
Титровану кислотність виноградних вин та сусел прийнято визначати у
грамах виної кислоти (формула 8.5), а плодово-ягідних – яблучної (формула 8.6).
Результати паралельних визначень виражають з точністю до 0,01, а
кінцевий результат округлюють до 0,1.
8.3 Визначення вмісту вільних органічних
кислот
Суму вільних органічних кислот можна визначити йодометричним методом, рекомендованим Х. Н. Починком. При цьому відбувається наступна реакція:
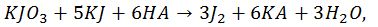
де А - аніон
кислоти.
Метод поширюється на свіжі фрукти, ягоди та овочі, а також іншу рослинну
сировину.
Суть методу
До розчину, який містить вільні кислоти, додають надлишок нейтрального розчину йодид-калій йодатом. Йод, який виділився титрують розчином гіпосульфіту. При цьому відбувається наступна реакція:
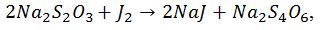
Ця реакція в розчинах з високою концентрацією іонів водню проходить дуже
швидко, але при зниженій концентрації іонів водню її швидкість сповільнюється,
тому слабкі кислоти цим методом безпосередньо визначати не можна, так як
концентрація іонів водню в кінці титрування недостатня для перебігу
реакції.
Аніони органічних оксикислот утворюють комплексні сполуки з іонами
кальцію, барію і магнію і в присутності цих катіонів поводяться як сильні
кислоти. Щавлева кислота, на відміну від ряду інших
органічних кислот, сильно дисоційована, і її також можна визначати цим методом. Винну, лимонну, яблучну і молочну
кислоти можна визначити кількісно, якщо до розчину цих кислот
додати хлористий барій або хлористий кальцій, а потім йодид і йодат калію.
Приблизно через 30 хв реакція закінчується і йод, який виділився, відтитровують
гипосульфітом. Бензойну і бурштинову кислоти цим методом визначити не можна, так
як вони не утворюють міцних комплексних сполук з іонами лужноземельних металів.
Однак вміст цих кислот в рослинах зазвичай дуже низький і не перевищує сотих
часток відсотка, тому при визначеннях суми вільних органічних кислот їх
кількістю можна знехтувати.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали: центрифуга, ступки порцелянові, ковби мірні на 50 см3, піпетки, бюретки; 10% -й розчин ВаС12, 0,01 н розчин гіпосульфіту, 1% -й розчин крохмалю, 0,1 н йодид-йодатний розчин: 3,567 г KJO3 і 40 г KI розчиняють у воді, доводять об'єм розчину до 1 л, зберігають у темній склянці.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Хід аналізу:
1. Наважку свіжого рослинного матеріалу (5…10 г) переносять у порцелянову
ступку і розтирають з 5 см3 10 % -го розчину хлористого барію;
2. Розтерту масу переносять у центрифужну пробірку;
3. Ступку ретельно змивають невеликими порціями дистильованої води і всі
промивні води додають у центрифужну пробірку;
4. Центрифугують 3…5 хв при 2…3 тис. об / хв;
5. Надосадову рідину зливають в мірну ковбу місткістю 50
см3;
6. До осаду в центрифужній пробірці доливають 1 см3 10 % -го
розчину хлористого барію, 8…10 см3 дистильованої води і знову
центрифугують;
7. Надосадову рідину зливають в мірну ковбу;
8. Осад ще раз промивають дистильованою водою, центрифугують і надосадову
рідину знову зливають в мірну ковбу;
9. об'єм розчину в мірній ковбі доводять дистильованою водою до 50
см3 і перемішують;
10. Беруть піпеткою 10 см3 розчину, переносять в конічну ковбу для
титрування;
11. Додають в ковбу 5 см3 10 %-го розчину хлористого барію і 5
см3 0,1 н йодид-йодатного розчину;
12. Перемішують, закривають ковбу пробкою і залишають на 30
хв;
13. Після цього додають 0,5 см3 розчину крохмалю і титрують 0,001 н розчином гіпосульфіту до зникнення синього забарвлення.
Розрахунки:
Вміст вільних кислот у досліджуваному матеріалі в перерахунку на яблучну кислоту розраховують за формулою:

де Х - вміст вільних кислот у
перерахунку на яблучну кислоту,%;
0,067 - 1 мекв яблучної кислоти;
50 - загальний об'єм досліджуваного
розчину, см3;
Т - поправка до титру 0,01 н.
розчину гіпосульфіту;
а - об'єм 0,01 н. розчину гіпосульфіту,
витрачений на титрування, см3;
100 - коефіцієнт для перерахунку в
відсотки;
10 - об'єм досліджуваного розчину,
взятий для титрування, см3;
М – маса наважки свіжого або абсолютно
сухого аналізованого рослинного матеріалу, г.
За результат вимірювань приймають середньоарифметичне значення двох паралельних визначень, округлене до сотих часток.
8.4 Визначення вмісту летких
кислот
Метод поширюється на продукти переробки фруктів і овочів, фруктові та
овочеві соки, нектари, морси та соковмісні напої, фруктові та овочеві
концентровані соки, пюре та концентровані пюре, морси і концентровані морси,
компоти, киселі, в тому числі виготовлені із сушених фруктів (сухофруктів),
джеми, повидло, варення (далі - продукти) і дає змогу встановлювати масову
частку летких кислот (у перерахунку на оцтову кислоту). Діапазон вимірювання
масових часток летких кислот від 4•10-2% до 1%. Межа виявлення методу
2•10-2%.
Суть методу
Полягає у виділенні летких кислот оцтовокислого ряду (оцтової,
пропіонової, тощо) шляхом
відгону з водяною парою та титруванні
отриманого дистиляту розчином гідроксиду натру в присутності фенолфталеїну
у якості
індикатора.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: терези неавтоматичної дії II класу
точності з найбільшою межею зважування 500 г і ціною повірочної поділки 0,01 г;
мікробюретки місткістю 10 см3 з ціною поділки 0,02 см; піпетки з
однієї міткою 2-2-2, 2-2-10, 2-2-20 і 2-2-100; склянки В-1-100; лійки скляні;
ковби мірні 2-1000-2; електроплитки побутові; ковбонагрівач електричний з
регулюванням температури; секундомір.
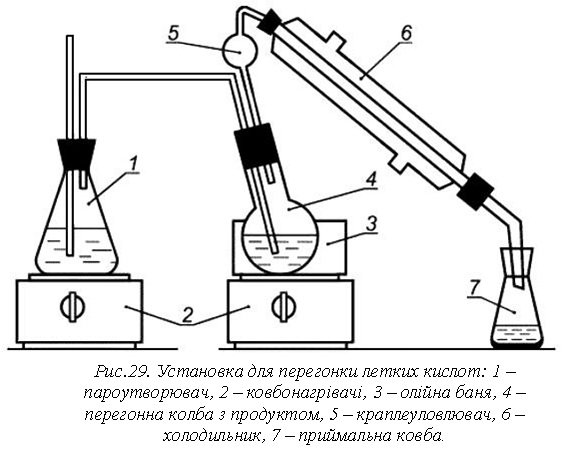
Установка для перегонки летких кислот (рис. 29) складається з ковби
круглодонної місткістю 1000 см3, ковби круглодонної з двома
горловинами з шліфами, краплевловлювача з відведенням типу КО-4, холодильника
спірального з внутрішнім охолодженням ХСВ або прямого ХПТ з довжиною кожуху 400
мм, ковби конічної, трубки поєднувальної скляної, діаметром 4 мм, трубки
поєднувальної гумової.
Допоміжні матеріали та реактиви: шматочки порцеляни або пемзи; кислота оцтова льодяна х.ч., натру
гідроокис х.ч. або стандарт-титр, крохмаль розчинний ч.д.а., фенолфталеїн
ч.д.а., спирт етиловий ректифікований, вода для лабораторного аналізу 2-го
ступеня чистоти.
Допускається застосування інших засобів вимірювальної техніки, допоміжного обладнання, які не поступаються вищезазначеним за метрологічними і технічними характеристиками, а також реактивів, посуду і матеріалів, за якістю не гірше вищезазначенних.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні.
Приготування розчинів:
- приготування розчину гідроокису натру молярної концентрації
0,1 моль / дм3: для приготування розчину гідроокису натру молярної концентрації 0,1
моль / дм3 вміст ампули стандарт-титру кількісно переносять
в мірну ковбу на 1000 см3, доводять об'єм розчину до мітки водою і
перемішують. Розчин зберігають в закритому посуді з полімерного матеріалу не
більше 6 місяців за умови перевірки його концентрації не менше одного разу на
місяць. При наявності осаду або помутніння розчин не
використовується.
- приготування розчину гідроокису натру молярної концентрації 0,01 моль / дм3: у мірну ковбу місткістю 1000 см3 вносять (100 ± 1) см3 розчину гідроокису натру молярної концентрації 0,1 моль / дм3, доводять об'єм розчину до мітки дистильованою водою, яка не містить вуглекислоти, і перемішують. Розчин використовують свіжоприготовленим.
Підготовка установки:
- збирають установку для перегонки летких кислот відповідно до креслення,
наведеному на рисунку 29;
- пар, який надходить з пароутворювача, не повинен містити вуглекислого
газу. При додаванні до 200 см3 водного дистиляту 0,1 см3
розчину гідроокису натру молярної концентрації 0,01
моль / дм3 у присутності двох крапель розчину фенолфталеїну
повинно з'явитися рожеве забарвлення, яке не зникає протягом 10
с.
- критерієм придатності установки для перегонки летких кислот є виявлення
не менше 99% оцтової кислоти в дистилляті, отриманому при перегонці 20
см3 атестованого розчину оцтової кислоти з масовою часткою 1% (див.
додаток Б, таблиця Б 2);
- після кожного визначення установку для перегонки летких кислот ретельно
промивають гарячою водою і висушують;
- деталі установки з'єднують між собою за допомогою конічних
взаємозамінних шлифов і закріплюють пружинами;
- допускається застосування установок інших типів, що відповідають вимогам, наведеним вище.
Хід аналізу:
Перегонка:
1. У хімічному стакані зважують від 25 до 50 г рідких або від 10 до 15 г
густих і в'язких продуктів, попередньо підготовлених;
2. Продукт з хімічного склянки кількісно переносять в перегінну ковбу,
змиваючи стакан водою для лабораторного аналізу, взятої в такій кількості, щоб
загальний об'єм суміші в ковбі склав близько 100
см3;
3. Пароутворювач зі шматочками фарфору або скляними кульками, для
забезпечення рівномірного кипіння, наповнюють на 2/3 об'єму водою:
4. Воду доводять до кипіння і протягом 10 хв. через всю установку
пропускають пар при відключеному струмі води через
холодильник;
5. Подають воду в холодильник установки, одночасно зменшуючи подачу пара з
пароутворювача на час внесення проби продукту в перегінну ковбу;
6. Вміст перегінної ковби нагрівають до кипіння і відганяють леткі кислоти;
7. У процесі перегонки нагріванням регулюють надходження пара з ковби
пароутворювача, забезпечуючи рівномірне проходження його через продукт так, щоб
об'єм аналізованої проби був постійним і становив приблизно 150
см3;
8. Перегонку закінчують після отримання в приймальній ковбі 200
см3 відгону.
9. У разі необхідності послаблюють затиск паровідвідних труб, щоб випустити частину пара в повітря.
Титрування:
10. До отриманого дистиляту додають дві-три краплі розчину фенолфталеїну;
11. Титрують з бюретки розчином гідроокису натру до появи світло-рожевого
забарвлення, яке не зникає протягом 30 с;
12. Проводять два паралельних визначення та записують об'єм розчину гідроокису натру, який витрачено на титрування.
Розрахунки:
1. Масову частку летких кислот (в перерахунку на оцтову кислоту) в продукті Х1,%, обчислюють за формулою:

де 60 - молярна маса оцтової кислоти,
г / моль;
С - молярна концентрація розчину гідроокису натру,
моль / дм3;
V – об'єм розчину гідроокису натру, витраченого на титрування дистиляту,
см3;
m - маса продукту, взятого на визначення, г;
0,1 - коефіцієнт перерахунку.
2. При вмісті в продукті загального двооксиду сірки понад 0,01% за масою, масову частку летких кислот (в перерахунку на оцтову кислоту) в продукті, %, обчислюють з урахуванням поправки за формулою:

де Х0
– масова частка загального двооксиду сірки в продукті,
%.
За результат вимірювань приймають середньоарифметичне значення двох паралельних визначень. Всі обчислення проводять до третього десяткового знака.
8.5 Визначення вмісту l–яблучної кислоти
ферментативним методом
Метод поширюється на фруктові і овочеві соки, в тому числі
концентровані, нектари, соковмісні напої, пюре і концентровані пюре, морси і
концентровані морси (далі - сокова продукція). Діапазон
вимірювань масової концентрації L-яблучної кислоти від 0,01 до 10,0
г / дм3
включно.
Суть методу
Полягає у
ферментативному перетворенні L-яблучної кислоти в іон оксалоацетату
під дією НАД у
присутності L-МДГ, зміщенні рівнотерези реакції шляхом зв'язування утвореного іона
оксалоацетата L-глутаматом в
присутності ГОТ, з подальшим спектрофотометричним
вимірюванням
кількості утвореного
НАДН, яка
еквівалентна вмісту
L-яблучної кислоти.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: терези; спектрофотометр; кювети з оптичного скла або полімерні з довжиною
оптичного шляху 10 мм; іономір або рН-метр в комплекті з комбінованими
мікроелектродами для вимірювань в розчинах об'ємом не менше 2 см3;
дозатори лабораторні об'ємом дозування 1, 0,2 і 0,02 см3; лійки
лабораторні; фільтри мембранні, діаметром 13 або 47 мм; фільтри паперові;
центрифуга лабораторна; мішалка магнітна з кутовою швидкістю обертання від 400
до 1200 хв.; шпателі пластикові або палички скляні оплавлені довжиною від 2 до 5
см; пробірки центрифужні з полімерного матеріалу місткістю 15 або 50
см3 з кришками; електроплитка лабораторна.
Набір реагентів, який включає:
- реактив 1 - флакон, що містить суміш гліцилгліцинового буферного розчину, рН 10,0 і L-глутамінової кислоти: 4,75 г гліцилгліціна і 0,88 г
L-глутамінової кислоти розчиняють в 50 см3 дистильованої
води. Встановлюють активну кислотність розчину на рівні 10,0 pH приблизно
4,6 см3 розчину гідроксиду натру молярної концентрації
(NaOH) = 10 моль / дм3. Об'єм розчину доводять дистильованою водою до 60
см3. Буферний розчин стійкий за температури 4 ° С 3 місяці;
- реактив 2
- флакон, що містить ліофілізат НАД: 0,420 г НАД розчиняють в 12 см3
дистильованої води. Розчин стійкий за температури 4 ° С 1
місяць;
- реактив 3
- суспензія ГОТ активністю 160 Е: розчин глутаматоксалоацетаттрансамінази
масової концентрації 0,002 г / см3, який містить L-аспартат
і кетоглутарат як субстрат, змішують з розчином Сульфатнокислого амонію молярної
концентрації (NH4)2SO4= 3,2 моль / дм3. Питома активність суспензії ГОТ становить не менше 400 Е/см3 Суспензія стійка за температури
4 ° С 12 міс.
- реактив 4
- суспензія L-МДГ активністю 2400 Е: розчин L-малатдегідрогенази масової
концентрації 0,005 г/см3 змішують з розчином сульфатнокислого амонію
молярної концентрації (NH4)2SO4 = 3,2 моль / дм. Питома активність суспензії L-МДГ становить
не менше 6000 Е / см3. Суспензія стійка за температури 4
° С 12 місяців;
- реактив 5
- контрольний розчин L-яблучної кислоти.
Полівінілполіпірролідон (ПВПП) низькомолекулярний або поліамід
(ПА).
Вода для лабораторного аналізу не нижче 2-го ступеня
чистоти.
Допускаються до застосування готові набори для визначення
L-яблучної кислоти фірми R-Biopharm AG, а також інші засоби
вимірювальної техніки, допоміжного обладнання, які не поступаються
вищезазначеним за метрологічними і технічними характеристиками, а також посуд,
матеріали і реактиви, за якістю не нижче вищезазначених.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Підговка проб:
Продукти зі стабільною каламутною
суспензією перед розведенням ретельно перемішують і центрифугують за швидкості
обертання 4000 хв-1 протягом 10 хв і фільтрують через паперовий або
мембранний фільтр з розміром пор 0,45 мкм.
Забарвлену сокову продукцію освітлюють шляхом додавання ПА або ПВПП. У центрифужну пробірку
місткістю 15 або 50 см3 додають до 10 см3 проби, близько
0,1 г поліаміду (ПА) або полівінілпіролідону (ПВПП), потім суміш інтенсивно
струшують в пробірці протягом 1 хв, центрифугують при швидкості обертання 4000
хв-1 протягом 10 хв і фільтрують через паперовий або мембранний
фільтр з розміром пор 0,45 мкм. Для визначення використовують прозорий
фільтрат.
Концентровану сокову продукцію розводять водою до отримання значення відносної густини розведеного продукту згідно
рецептури натурального соку, нектару або соковмісного напою. При цьому відносну
густину розведеної проби виражають в грамах на кубічний дециметр. Масову частку сухих розчинних речовин визначають за рефрактометром. При
розрахунках результатів випробувань враховують масу проби концентрованого
продукту і фактор розведення.
Пробу розводять дистильованою водою до отримання значення масової концентрації L-яблучної кислоти в
інтервалі від 0,02 до 0,35 г/дм3.
Приготування розчинів реактивів з готового набору проводять відповідно до інструкції, що додається до
набору.
Хід аналізу:
Приготування контрольного розчину:
- у фотометричну кювету вносять 1 см3 реактиву 1;
- 0,2 см3 реактиву
2;
- 1,5 см3 дистильованої води;
- 0,01 см3 реактиву
3;
- суміш ретельно перемішують пластиковим шпателем або скляною паличкою,
витримують 3 хв.;
- вимірюють оптичну густину розчину відносно оптичної густини повітря (А1)к.
Приготування дослідного розчину проби:
- у фотометричну кювету вносять 1 см3 реактиву 1;
- 0,2 см3 реактиву
2;
- 1,4 см3 дистильованої води;
- 0,01 см3 реактиву
3;
- 0,1 см3 підготовленої проби;
- суміш ретельно перемішують пластиковим шпателем або скляною паличкою,
витримують 3 хв.;
- вимірюють оптичну густину розчину відносно оптичної густини повітря (А1)д.
Ферментативна реакція та кількісне визначення:
- в кожну з кювет з контрольним та дослідним розчином додають по 0,01
см3 реактиву 4;
- суміш ретельно перемішують пластиковим шпателем або скляною паличкою,
витримують 5…10 хв.;
- вимірюють оптичну густину розчинів відносно оптичної густини повітря (А2)д і (А2)к;
- завершення реакції перевіряють виміром оптичної густини повторним
визначенням через 2 хв, а далі, за потреби кожні 5 хв. протягом 30 хв., щоб
встановити час початку збільшення значення оптичної щільності на постійну
величину.
Розрахунки:
В ферментативних реакціях, які лежать в основі даного методу, НАДН який
утворюється в кюветі призводить до зміни оптичної густини розчину ΔА, а його масова концентрація
пропорційна масовій концентрації L-яблучної кислоти в
пробі.
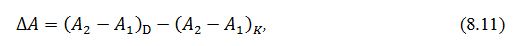
Масову концентрацію L-яблучної кислоти в пробі ρ, г/см3,
визначають відповідно до закону Бугера – Ламберта – Бера за формулою:

де М1 – молярна
маса L-яблучної кислоти, 134,09 г/моль;
V1
–
загальний об'єм розчину в кюветі, см3;
F – фактор розбавлення;
δ
– товщина поглинаючого шару в кюветі, см;
V2
–
об'єм проби, см3.
У випадку точного приготування проб (як зазначено вище) формула 8.12
приймає вигляд:

При
застосуванні готових наборів реактивів числовий коефіцієнт (3,647) у рівнянні
8.6 може мати інше значення, що пов'язано зі зміною загального об'єму
розчину V1
в кюветі.
За результат вимірювань приймають середньоарифметичне значення двох паралельних визначень, округлене до сотих часток.
8.6 Визначення вмісту лимонної
кислоти
Лимонна кислота С6Н8О7 є
одним з компонентів циклу трикарбонових кислот.
Вона накопичується в значних кількостях в плодах і листях. У
плодах цитрусових, ягодах журавлини і інших культурах її вміст досягає більше
90% кількості всіх кислот. Метод поширюється на свіжу плодово-ягідну та овочеву
сировину, а також на продукти її переробки.
Суть методу
Полягає у окисленні
лимонної кислоти перманганатом калію до
ацетондикарбонової
кислоти і бромуванні
цього продукту до появи
пентабромацетону.
Останній визначають ваговим
або
об'ємним методом.
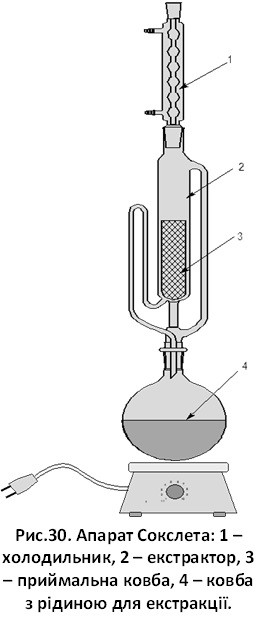
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали:
терези спеціального (I) класу точності з межами абсолютної похибки однократного зважування ±
0,001 г; фільтри мембранні; фільтри паперові; центрифуга лабораторна; мішалка
магнітна з кутовою швидкістю обертання від 400 до 1200 хв; ступка порцелянова;
циліндри мірні; колби конічні або круглодонні на 100 та 250 см3;
ексикатор; бюретки, піпетки, сушільна шафа або баня повітряна (комбінована);
апарат Сокслета (рис. 30).
Реактиви: 20 % сульфатна кислота; 20 % розчин бромистого калію;5 %розчин перманганату;
Насичений розчин сульфатнокислого заліза (40 г в 100 см3
води, яка підкислена сульфатною кислотою); 0,1 н розчин їдкого натру або їдкого
калію; 5% -вий розчин фосфорновольфрамової кислоти, папір фільтрувальний.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні.
Хід аналізу:
1.Вилучення лимонної кислоти:
- Беруть 5…10 г наважки (залежно від кількості лимонної кислоти в сировині),
- Наважку ретельно розтирають у ступці з додаванням товченого скла;
- Переносять розтерту наважку без втрат у мірний циліндр на 250 см3 і водою доводять до 100 см3;
- Додають 15 см3 20 % - вої сульфатної кислоти (1:8);
- Залишають на 15…30 хвилин;
- Доводять дистильованою водою до 200 см3;
- Суміш добре струшують і залишають на 2 години;
- Протягом настоювання вміст ковби неодноразово збовтують;
- Після настоювання додають 5…10 см3 5 % -вої фосфорновольфрамової або метафосфорної кислоти;
- Добре перемішують і центрифугують або фільтрують через сухий фільтр у
сухий посуд.
Добрим
розчинником лимонної кислоти є ацетон
(в
100 частинах розчинника 4 частини кислоти). Вилучення органічних
кислот ацетоном з сухого матеріалу виконують у апараті
Сокслета (рис. 30).
Ацетонові витяжки спрощують
подальші
процедури.
2.Окиснення
фільтрату
- 50 см3 прозорого фільтрату наливають у колбу на 200
см3;
- Додають 5 см3 30 %-вого розчину бромистого
калію;
- 10 см3 розведеної сульфатної кислоти (1:1);
- Після перемішування швидко додають 20 см3 5 % -вого
розчину перманганату калію;
- Сміш
залишають на 10 хв. у витяжній шафі під
тягою,
періодично
збовтуючи без нагрівання;
- Потім
охолоджують
колбу
до температури 10 °С;
- Якщо
бурий
осад двоокису мангану, який з'явився,
зникає, то визначення треба
повторити
з додаванням
більшої кількості розчину перманганата;
- Надлишок
окислювача видаляють додаванням 20 см3 насиченого
розчину
закисного
Сульфатнокислого заліза до повного
знебарвлення;
- Суміш
з утвореним
пентабромацетоном
ставлять
у воду із льодом або в холодильник;
- Суміш у холодильнику залишають до ранку. За
цей час
мутний розчин поступово освітлюється, а осад ущільнюється
на
дні колби;
- Потім
зливають рідину з осаду декантацією через скляний
мембранний фільтр (або тигель
з пористим дном);
- Фільтр
з осадом ретельно
промивають
до нейтральної реакції за
метилоранжем: осад
пентабромацетона повинен бути чисто білим або слабо
забарвленим;
- Фільтр з осадом висушують в ексикаторі над сульфатною
кислотою;
- Зважують і, знаючи вагу сухого фільтра, за різницею встановлюють вагу
осаду пентабромацетона (СНВr2–СО – СВг3), 1 мг якого відповідає 0,483 мг лимонної
кислоти;
Кількість
пентабромацетона можна визначити об'ємним методом.
В цьому випадку:
- Промитий
осад з фільтра переносять в колбу на 200 см3, розчиняючи в
20 см3
спирту;
- Додають
50 см3 0,1 н розчину лугу;
- Колбу ставлять у сушильну шафу (або повітряну баню (рис. 31));

- Нагрівають
30 хвилин за
температури
не вище
90 °С
для повного розчинення осаду;
- Розчин
охолоджують;
- Надлишок
лугу відтитровують
0,1 н розчином сульфатної
кислоти
в присутності
метилового червоного
до появи яскраво-рожевого
забарвлення.
Розрахунки:
Кількість лимонної кислоти у відсотках (Х) обчислюють за формулою:

де:
а –
кількість 0,1 н лугу,
пов'язаного в
реакції з пентабромацетоном, см3;
0,483 - кількість лимонної кислоти, яка відповідає 1 см3 0,1 н
лугу, мг;
К
–
коефіцієнт
розведення (відношення об’єму
екстракту
до
об’єму,
який взято
для окиснення);
т –
маса наважки
матеріалу, г.
За результат вимірювань приймають середньоарифметичне значення двох
паралельних визначень, округлене до сотих часток.
8.7 Визначення вмісту бурштинової
кислоти
В незрілих плодах та паростках деяких рослин міститься бурштинова кислота ( СООН-СН2-СН2-СООН) в
значних кількостях.
Метод поширюється на різні види рослинної сировини, у тому ж разі
на плоди та овочі.
Суть методу
Полягає у вилученні органічних
кислот та переведенні
їх у
барієві солі. Бурштинову кислоту після окислення
перманганатом калію
відокремлюють від інших органічних кислот (лимонної,
яблучної)
і титрують.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали:
терези спеціального (I) класу точності з межами абсолютної похибки однократного зважування ±
0,001 г; фільтри мембранні; фільтри паперові; центрифуга лабораторна; мішалка
магнітна з кутовою швидкістю обертання від 400 до 1200 хв; ступка порцелянова;
циліндри мірні; колби конічні або круглодонні на 100 та 250 см3;
ексикатор; бюретки, піпетки, сушільна шафа; водяна баня; апарат Сокслета (рис.
30).
Реактиви: розчин сульфатної кислоти (1:4); безводний сульфатнокислий натр; їдкий
барій; 10 % -вий розчин хлористого барію; 5 % -вий розчин
перманганату калію; 0,1 н розчин їдкого натру або їдкого калію; етиловий етер і
спирт.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Хід аналізу:
1.Вилучення кислот:
- Наважку близько 10 г розтирають із 4 см3 розчину сульфатної
кислоти (1:4);
- Далі зневоднюють 5 г безводного Na2S04 і органічні кислоти вилучають сухим сульфатним етером у
апараті Сокслета (можна замінити це вилучення струшуванням з етером, так як
бурштинова кислота добре розчиняється в етері);
- Етерний
екстракт переливають цілком у
ділільну
лійку на 250 см3;
- Збовтують
із
100 см3 дистильованої води (20…30
хвилин) до повного вилучення органічних
кислот;
- Етер
при цьому знебарвлюється, а водна витяжка забарвлюється, так як в воду, крім
органічних кислот, переходять пігменти і деякі смоли;
- Водний
шар з ділильної лійки
зливають в мірну колбу на 250 см3 і доводять водою до риски;
- З
цього вихідного розчину беруть окремо пробу для визначення
бурштинової
кислоти;
- Залишки
етеру
видаляють випаровуванням;
- Об'єм рідини доводять дистильованою водою до 100 або 200 см3.
2.Отримання барієвих солей органічних кислот.
- Беруть 50…100 см3 охолодженої витяжки в круглодонну колбу на
200 см3;
- Нейтралізують 1 н розчином їдкого барію;
- Додають 1 см3 10 % - вого розчину хлористого
барію;
- Колбу нагрівають 10 хв. на киплячій водяній бані зі зворотним
холодильником;
- Вміст колби переносять в порцелянову чашку і випарюють на водяній бані до
утворення стану сиропу;
- Залишок після випарювання розчиняють у 20 см3
води;
- Додають 80 см3 95%-вого спирту;
- Ретельно перемішують та настоюють 1…2 години.
3.Розділення органічних кислот:
- Після закінчення зазначеного часу витримки осад відсмоктують і промивають
спиртом;
- Потім осад кількісно переносять у колбу, змивають з фільтра струменем
дистильованої води;
- Після додавання в цілому близько 70 см3 води колбу нагрівають
для видалення спирту;
- Потім, колбу нагрівають та поступово додають 5 % -вий розчин
перманганату калію до стійкого збереження червоного
кольору;
- Надлишок перманганату калію і осад перекису мангану видаляють додаванням
3 % - вого гідроген пероксиду або додаванням сульфіту після
підкислення реактивної суміші сульфатною кислотою;
- Бурштинову кислоту відокремлюють етиловим етером в апараті Сокслета
протягом 8 годин з сухого порошку, який отримано після випарювання водного
розчину до 10-15 см3 з подальшим змішуванням з 5 г безводного
сульфатнокислого натру;
- Після вилучення етер відганяють;
- Доливають 20 см3 води;
- Титрують бурштинову кислоту 0,1 н лугом (1 см3 0,1 н. лугу
відповідає 5,9 мг бурштинової кислоти).
За результат вимірювань приймають середньоарифметичне значення двох паралельних визначень, округлене до сотих часток.
8.8 Визначення вмісту винної
кислоти
Винна
кислота [СООН - СН (ОН) – СН (ОН) - СООН]
у
великих кількостях знаходиться в
рослинах родин виноградних, геранієвих і бобових. З чотирьох стереоізомеров
винної кислоти в рослинах зазвичай зустрічається правообертаюча
форма.
Суть методу
Полягає у
осадженні винної кислоти в присутності оцтової кислоти у вигляді важкорозчинної
кислої винно-калієвої
солі,
яку відтитровують
лугом:
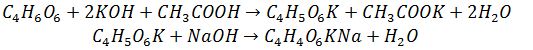
Метод
дозволяє кількісно визначити винну кислоту
в присутності інших органічних кислот.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: колба Бунзена, лійка Бюхнера, тиглі Гуча, насос Комовського,
терези; фільтри мембранні; фільтри паперові; центрифуга лабораторна; мішалка
магнітна з кутовою швидкістю обертання від 400 до 1200 хв.; ступка порцелянова;
циліндри мірні; колби конічні або круглодонні на 100 та 250 см3;
ексикатор; бюретки, піпетки, сушільна шафа; водяна баня; апарат Сокслета (рис.
30).
Реактиви:
1 н
розчин сульфатної кислоти; 1
н розчин їдкого
калію;
5% -вий
розчин фосфорновольфрамової
кислоти; етанол; 30
% -ва
оцтова кислота; 0,1 н розчин
їдкого натру, титр якого точно встановлений.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні.
Хід аналізу:
1.Вилучення кислот:
- наважку близько 10 г розтирають з 4 см3 розчину сульфатної
кислоти (1:4);
- зневоднюють 5 г безводного Na2S04 ;
- органічні кислоти витягають сухим сульфатним етером в апараті
Сокслета;
- етерний
екстракт переливають цілком у
ділільну
лійку на 250 см3;
- збовтують
із
100 см3 дистильованої води (20…30 хв.)
до повного вилучення органічних
кислот;
- етер
при цьому знебарвлюється, а водна витяжка забарвлюється, так як в воду, крім
органічних кислот, переходять пігменти і деякі смоли;
- водний
шар з ділильної лійки
зливають в мірну колбу на 250 см3 і доводять водою до риски;
- з
цього вихідного розчину беруть окремо пробу для визначення винної
кислоти;
- залишки
етеру
видаляють випаровуванням;
- об'єм рідини доводять дистильованою водою до 100 або 200 см3
2.Попереднє визначення вмісту огранічних кислот у водній
витяжці:
- витяжку титрують 0,1 н розчином лугу;
- виражають у кількості винної кислоти: 1 см3 0,1 н лугу відповідає 7,5 мг винної кислоти.
3.Осадження винної кислоти
- у витяжку додають 5…10 см3 5 % -вої
фосфорновольфрамової або метафосфорної кислоти;
- добре перемішують і центрифугують або фільтрують через сухий фільтр у
сухий посуд.
- об'єм витяжки повинен містити не менше 100 мг винної
кислоти;
- витяжку випарюють до 20 см3 у хімічній склянці на 150
см3.
- після випарювання, витяжку охолоджують протягом 2…3 годин у
холодильнику;
- до охолодженої витяжки доливають (до нейтралізації за фенолфталеїном) 1
н розчин їдкого калію і ще надлишок в 3 краплі;
- додають 4 см3 80 % -вої оцтової
кислоти;
- протягом двох хвилин доливають 80 см3 95 % спирту при
постійному перемішуванні;
- розчин продовжують енергійно перемішувати скляною паличкою для
прискорення випадання осаду;
- при уповільненому
осадженні труть
паличкою
об стінку склянки;
- склянку з осадом
білих
кристалів
кислої винно-калієвої солі залишають
на ніч у холодильнику;
- далі
спирт зливають декантацією;
- осад, змиваючи 80 % - вим
охолодженим спиртом,
поступово
переносять
в тигель Гуча або стаканчик з пористим дном, застосовуючи
відсмоктування;
- осад
промивають
до
зникнення кислої реакції в промивному
спирті
(за
фенолфталеїном). Триразового
промивання по 15 см3
зазвичай буває достатньо;
- промитий
осад з тигля разом з азбестом переносять
назад
в ту
ж склянку
на 150 см3
за допомогою 100 см3 гарячої води;
- для кращого розчинення осаду вміст ретельно
збовтують;
- стакан з розчином нагрівають майже до кипіння;
- кислий виннокислий калій, який повністю розчинився відтитровують 0,1 н лугом з фенолфталеїном (1 см3 0,1 н. розчину їдкого натру, який пішов на титрування цієї солі, відповідає 15 мг винної кислоти).
Розрахунки:
Вміст
винної кислоти у відсотках (Х)
обчислюють за
формулою:

де:
0,015 г винної кислоти, які відповідають
1 см3 0,1 н
розчину
їдкого
натру;
а
– кількість 0,1 н розчину їдкого натру,
який
пішов на титрування кислого виннокислого калію,
см3;
V –
об’єм
витяжки, см3;
V1 –
об’єм
проби, взятий
для аналізу для осадження винної
кислоти, см3;
m
–
маса наважки
матеріалу, г.
Приклад розрахунку. Наважка свіжих ягід винограду 100 г доведена до 500 см3 звідки взято 100 см3 для осадження винної кислоти. На титрування осаду кислого виннокислого калію пішло 22,7 см3 0,1 н NaOH. Отже, відсоток вільної та зв'язаної винної кислоти (Х), %:
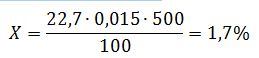
За результат вимірювань приймають середньоарифметичне значення двох паралельних визначень, округлене до сотих часток.
8.9 Визначення вмісту щавлевої кислоти
У
формі слаборозчинної
солі кальцію, а також інших розчинних солей щавлева кислота (СООН - СООН)
широко поширена в рослинах. У вільному вигляді вона накопичується в значних
кількостях у
старіючих листках щавлю, шпинату, буряка, черешках листя ревеню та інших
рослин.
Суть методу
Полягає у
осадженні щавлевої кислоти у вигляді щавлевокислого кальцію, майже нерозчинного
в холодній воді.
Метод
дозволяє визначати від 3 до 200 мкг щавлевої кислоти в пробі.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали:
колба Бунзена, тиглі Гуча, насос Комовського, терези; фільтри мембранні; фільтри паперові; центрифуга лабораторна;
гомогенізатор; мішалка магнітна з кутовою швидкістю обертання від 400 до 1200
хв.; ступка порцелянова; циліндри мірні; колби конічні або круглодонні на 100 та
250 см3; ексикатор; бюретки, піпетки, сушільна шафа; водяна баня;
апарат Сокслета (рис. 30).
Реактиви: реактив для осадження щавлевої кислоти (до розчину, який складається з
25 г хлористого кальцію розчиненого в невеликій кількості води та налитого в
мірну колбу на 500 см3, доливають до мітки 50 % -вой
оцтовой кислотой і розчин 330 г кристалічного натрій ацетату в 300
см3 води: обидва розчини добре перемішують, отримують реактив та
витримують 48 годин за температури 5…7 °С і фільтрують); борна кислота; 0,1 н
розчин перманганату калію; 1 % -вий розчин AgNO3.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні.
Хід аналізу:
1.Вилучення кислот:
- наважку свіжого матеріалу 10 г розтирають з 4 г безводного
сульфату натру;
- витримують в термостаті при 40 ° С до появи кристалічної маси,
з якої витягають органічні кислоти;
- після 30 хв. струшування суспензію фільтрують через шар безводного
сульфату натру;
- шар сульфату натру після фільтрування промивають невеликою порцією
суміші розчинників, застосовуючи відсмоктування водоструминним насосом;
- потім
все вливають в порцелянову чашку і випарюють розчинник на водяній бані
насухо;
- сухий
залишок в чашці розчиняють в 1 см3 дистильованої води.
2.При визначенні в сухому матеріалі:
- 5 г сухого листя розтирають зі скляним піском та з 5 см3
10 % -вої сульфатної кислоти і невеликою кількістю спирту;
- все
переносять в мірний циліндр або колбу і доливають водою до 250…500
см3;
- струшують
і залишають на кілька годин або на ніч;
3.Осадження щавлевої кислоти:
- в колби на 200 см3 беруть по 50 см3 витяжки,
приготовленої як зазначено вище;
- додають до лужної реакції амоніак і 1-2 г борної кислоти (останню
додають для ускладнення осадження солей винної кислоти та її оптичних
ізомерів);
- додають 10 см3 реактиву для осадження щавлевої кислоти і
залишають на 48 годин за температури не вище 7 °С;
- осад, що виділився, щавлевокислого кальцію відфільтровують і промивають гарячою водою до негативної реакції на хлор (відсутність осаду від додавання декількох крапель 1% -вого розчину азотнокислого срібла);
4.Визначення щавлевої кислоти виконують одним з наступних способів:
Перший
спосіб:
- осад щавлевокислого кальцію змивають з фільтра водою з промивалки в
колбу;
- відразу ж доливають гарячу 10 % -ву сульфатну кислоту через
той же фільтр, щоб розчинити сліди оксалату;
- осад в колбі при помішуванні переходить в розчин;
- при повільному розчиненні осаду колбу з рідиною нагрівають;
- фільтр промивають гарячою водою і розчин титрують 0,1 н розчином
перманганату калію до появи рожевого забарвлення;
- знаючи, скільки см3 перманганату калію йде на титрування взятої проби і знаючи титр перманганату калію за щавлевою кислотою, дізнаються кількість кислоти (в г). Відсоток щавлевої кислоти (Х) знаходять за формулою:
де: b -
загальний об’єм витяжки,
см3;
а - щавлева кислота в пробі
витяжки,
%;
b1 -
об’єм
проби,
см3;
m – маса
наважки
досліджуваного
зразка,
г.
Другий спосіб
- осад
щавлевокислого кальцію разом з фільтром переносять в
колбу;
- розчиняють в точному об’ємі титрованого розчину H2SO4;
- надлишок кислоти відтитровують 0,1 н розчином
лугу;
- 1 см3 0,1 н H2S04 дорівнює 4,5 мг щавлевої кислоти.
За результат вимірювань приймають середньоарифметичне значення двох паралельних визначень, округлене до сотих часток.
8.10 Визначення піровиноградної кислоти бісульфітним
методом (за С. Б. Мешковою та Є. С. Северином)
Суть методу
Базується на тому, що кислі солі KHSO3, NaHSO3 в кислому середовищі реагують з карбонільною групою піровиноградної
килоти ПВК з утворенням бісульфітної сполуки. Надлишок доданого бісульфіту потім звязують
йодом:
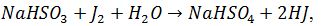
а бісульфітну похідну ПВК розкладають, додаючи до розчину, який вже не
має вільного бісульфіту, бікарбонат натру. Кількість звільненого при цьому
бісульфіту еквівалентна кількості ПВК. Відтитровуючи бісульфіт йодом по
кількості витраченого на титрування йоду, розраховують кількість ПВК.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали: мікробюретки (або бюретка) з поділами 0,025 см3, мірні колби на 200 та 300 см3; конічні колби; піпетки на 5, 10, 20 см3.
Реактиви: 1% розчин натрій бисульфиту (або калій, зберігають 2-3 дні); 0,1 і 0,01 н розчини йоду; натрій двовуглекислий; 1% розчин крохмалю; 5…10% розчин фосфорновольфрамовой кислоти;
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні.
Хід аналізу:
- у конічну колбу наливають 20…25 см3 водної витяжки
досліджуваного зразка;
- осаджують білки додаванням кількох крапель розчину фосфорновольфрамовой
кислоти;
- до 5…10 см3 витяжки, з якої осаджений білок
фосфорновольфрамовой кислотою, додають 3 см3 1% розчину натрій
бисульфиту;
- вміст колби добре перемішують і залишають на 30
хв.;
- потім надлишок бисульфиту видаляють спочатку 0,1 н розчином, а потім
0,01 н розчином йоду в присутності 1 см3 розчину крохмалю;
- хімічне з'єднання піровиноградної кислоти з бісульфітом руйнують
додаванням 1 г сухого натрій бікарбонату;
- виділений бісульфіт відтитровують 0,01 н розчином
йоду;
- визначення виконують із розрахунку: 1 см3 0,01 н розчину йоду
відповідає 0,44 мг піровиноградної кислоти.
За результат вимірювань приймають середньоарифметичне значення двох паралельних визначень, округлене до сотих часток.
8.11 Визначення вмісту молочної
кислоти
Молочна кислота знайдена у багатьох рослинах. Вона часто формується при
аеробному диханні, особливо у великій кількості - при молочнокислому бродінні.
Суть методу
Полягає в тому, що нагрівання молочної кислоти з концентрованою сульфатною кислотою веде до формування оцтового альдегіду, кількість якого визначають фотоме трично за реакцією з n-оксидифенілом в присутності іонів міді (Си 2+). Метод дозволяє визначити 2…10 мкг молочної кислоти в пробі. Крім молочної кислоти, цим методом можна визначити диоксиацетон, гліцериновий альдегід, n-оксифенілмолочну, яблучну та піровиноградну кислоти. Однак у відношенні молочної кислоти даний метод в 40…200 разів чутливіший. Присутність органічних кислот не заважає визначенню.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали: пробірки 20…30 мм з притертими пробками, водяна баня;
Реактиви: розчин CuSO4•5H2O (12 %); H2SO4 (густиною 1,84) яка не містить NO3- ; 1,5 %-й n-оксидифеніл у 0,1 н NAOH (1,5 г n-оксидифенілу розчиняють при нагріванні у 10 см3 1 н
NAOH, переносять в мірну колбу на 10 см3 й після охолодження
доливають до позначки водою, використовують протягом 1 місяця).
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні.
Хід аналізу:
- наважку 0,3…0,5 г екстрагують водою (100:1) 2 години при 70
°С;
- частину фільтрату, отриманого після екстракції наважки, наносять на
колонку (довжина 20 см, внутрішній діаметр 0,7 см) з катіонітом КУ – 2 у формі
Н+(І);
- після пропускання крізь катіоніт, фільтрат, який містить органічні
кислоти, цукри та інші неелектроліти, переносять на аніоніт ЕДЕ-10П у формі
ОН-;
- органічні кислоти затримуються на аніоніті, а цукри та основи
вилучаються з фільтратом та промивними водами;
- для вилучення зв'язаних з аніонітом кислот використовують 1 н розчин
Na2CO3;
- отриманий фільтрат, який містить
натрієві солі кислот, знову
пропускають крізь катіоніт (ІІ), в результаті чого формується елюат, до складу
якого входять вільні органічні кислоти;
- 1 см3 елюату наливають в пробірку з скляною притертою
пробкою;
- додають мікропіпеткою 0,05 см3 12 %-вого розчину CuSO4 та приливають по краплинах повільно 6 см3 концентрованої
H2SO4 з бюретки (пробірку тримають у воді з льодом для зменшення втрат
оцтового альдегіду, який формується при нагріванні, та струшують при додаванні
H2SO4);
- потім пробірку закривають пробкою та поміщають на 5 хв. в киплячу водяну
баню (або на 30 хв. при 60 °С);
- потім пробірки охолоджують до 10…15 °С;
- в охолоджену суміш піпеткою вносять 0,1 см3 лужного розчину
n-оксидифенілу;
- осад, що випав, швидко та рівномірно диспергують;
- потім пробірку поміщають на 30 хв. у водяну баню з температурою 30 °С,
зрідка струшуючи;
- далі її занурюють на 90 с в киплячу воду для вилучення надлишку
n-оксидифенілу;
- після цього охолоджують до кімнатної температури в холодній
воді;
- оптичну густину фіолетового розчину визначають (проти контрольної проби
з реактивами) на спектрофотометрі при довжині хвилі 570 нм;
- подальший розрахунок виконують згідно калібрувального графіка, який
складають в межах від 1 до 10 мкг молочної кислоти у пробі.
За результат вимірювань приймають середньоарифметичне значення двох
паралельних визначень, округлене до сотих часток.