13.ВИЗНАЧЕННЯ ВМІСТУ БІОЛОГІЧНО АКТИВНИХ РЕЧОВИН
13.1 Визначення вмісту
вітамінів
Вітаміни - низькомолекулярні органічні сполуки
різноманітної хімічної природи; це речовини, які забезпечують нормальне
протікання біохімічних і фізіологічних процесів у живих організмах. Вітаміни
відносяться до групи біологічно активних сполук, що регулюють обмін речовин у мінімальних концентраціях. Вони
тісно пов'язані з ферментами, більшість з них входять до складу активних груп
двокомпонентних ферментів.
Відсутність або недостатня кількість в їжі вітамінів призводять до
глибоких порушень обміну речовин і в кінцевому рахунку до авітамінозів і
гіповітамінозів.
Важливими постачальниками вітамінів для людини і тварин є рослини. Тому
визначення вітамінів у рослинах необхідно перш за все для оцінки харчової
цінності рослинних продуктів. Вміст вітамінів у порівнянні з
вуглеводами, білками і жирами в рослинах значно менше, тому для точного їх кількісного визначення необхідне суворе дотримання методики
аналізів.
13.1.1 Визначення вмісту аскорбінової
кислоти (вітаміну С)
Аскорбінова кислота (вітамін С) поширена як у рослинних, так і в
тваринних тканинах. Організм людини, мавп і морської свинки не здатний
синтезувати аскорбінову кислоту і повинен отримувати її в готовому вигляді з
їжею. Основними джерелами вітаміну С виступають зелені рослини, свіжі овочі і
фрукти; продукти тваринного походження містять незначні його кількості.
Середній вміст вітаміну С (в мг%): перець солодкий, хрін – 100…200,
капуста кольорова – 50…100, томати – 20…40, цибуля – 40…60, яблука – 5…30, вишня
– 5…15, смородина чорна – 100…400, шипшина – 1000…4000, кора калини – 70…80,
лист кропиви - 50, олія обліпихи - 250, листя горобини – 25…200, плоди
горобини – 40…200.
Аскорбінова кислота - вітамін, який в організмі бере активну участь у
різноманітних окисно-відновних процесах. Це обумовлено тим, що існує цей вітамін
у двох активних формах - власне аскорбінової кислоти і дегідроаскорбінової
кислоти. Нижче описаний метод визначення аскорбінової кислоти за
І. К. Муррі.
13.1.1.1 Визначення
масової частки аскорбінової кислоти за фарбою Тільманса
Суть методу
Заснована на редукуючих властивостях аскорбвнової кислоти. Під дією
аскорбінової кислоти розчин 2,6-дихлорфеноліндофенола (індикатор), який має синє
забарвлення, відновлюється в безбарвну сполуку. Аскорбінову кислоту вилучають із
рослин сумішшю 2 %
хлоридної і 2 % метафосфорної кислот і розчин титрують 2,6-дихлорфеноліндофенолом відомого титру до рожевого
забарвлення. За кількістю фарби, витраченої на титрування, розраховують
вміст аскорбінової кислоти. Хлоридна кислота вилучає
з рослинної тканини вільну і зв’язану аскорбінову кислоту в екстракт.
Метафосфорна кислота осаджує білки та підвищує стійкість аскорбінової кислоти в
екстракті.
Аналіз
від взяття
наважки до титрування слід провести
дуже
швидко, без перерви.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали: 2 мікробюретки з градуюванням на 0,01 см3, місткістю 2…5 см3; піпетки на 5, 10 і 100 см3; мірні колби на 100, 200, 1000 см3; конічні колби на 100…200 см3; мірні циліндри на 10 і 50 см3; хімічні стакани на 50…100 см3; лійки діаметром 100…110 мм; порцелянові ступки діаметром 100…120 мм; ножі з нержавіючої сталі; скляний порошок (чисте прозоре лабораторне скло, подрібнюють у ступці і обережно просівають крізь сито з отворами діаметром 1,0…1,5 мм).
Реактиви:
- Суміш
2 % хлоридної і 2 % метафосфорної кислот:
44,3 см3
концентрованої хлоридної
кислоти
густиною 1,19 г/см3 наливають у мірну колбу на 1000 см3,
у яку вже налито воду, потім
додають
20 г метафосфорної кислоти й доводять водою до 1000 см3 ;
- 2
% сульфатна кислота:
11,6
см3
концентрованої кислоти густиною 1,84 г/см3
розводять
водою до 1000 см3;
- аскорбінова
кислота,
кристалічна;
- калій йодид,
кристалічний;
- розчин
крохмалю (індикатор):
1 г розтертого розчинного крохмалю збовтують із
20
см3
холодної дистильованої води й виливають цю суміш у 80 см3
гарячої води. Потім
кип’ятять
3 хв. на електроплитці. Розчин фільтрують гарячим; для користування
придатний
протягом 3 діб (за зберігання в холодильнику).
- 0,001
н
розчин калій йодату
(KJO3):
на
аналітичних терезах зважують 0,3568 г
калій йодату,
висушеного протягом 2-х годин
за температури 102ºС, розчиняють у воді й
доводять
до 1000 см3.
Отриманий 0,01 н
розчин розбавляють у 10 разів і зберігають у добре
закритій
посудині в теплому місці.
- 0,001 н розчин 2,6-дихлорфеноліндофенолу (фарби): на чистому годинниковому склі зважують 60 мг сухої фарби, переносять у мірну колбу на 200 см3, додають 100…150 см3 теплої дистильованої води і 4…5 крапель 0,01 н лугу. Колбу ретельно збовтують 10 хв., потім доливають до мітки водою, ще раз збовтують і фільтрують крізь щільний фільтр у суху колбу. Розчин фарби готують перед використанням, ним можна користуватися протягом 3-х діб, за умови обов’язкового встановлення титру в день застосування. За зберігання в холодильнику розчин можна використовувати протягом 7-ми діб.
Підготовка до проведення вимірювань
- відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1. Проби, які надходять у лабораторію повинні бути без пошкоджень і
змін якості продукту при транспортуванні та зберіганні;
- при приготуванні наважки плоди та овочі необхідно подрібнювати ножами і
теркою з нержавіючих матеріалів і як можливо швидше;
- встановлюють титр
розчину 2,6-дихлорфеноліндифенолу за калій йодатом. Цей
метод
засновано на паралельному титруванні розчину аскорбінової кислоти фарбою і 0,001
н
розчином калій йодату.
Титр
фарби
легко розрахувати, з погляду на те, що
1 см3
0,001 н
розчину калій йодату еквівалентний
0,088 мг аскорбінової кислоти.
Для встановлення титру фарби розчиняють кілька кристалів (KJO3), близько 1,0…1,5 мг аскорбінової кислоти у 50 см3
2 %-вої сульфатної кислоти. Піпеткою
беруть 5 см3
цього розчину в невелику склянку й титрують фарбою з
мікробюретки
до рожевого забарвлення, яке не зникає протягом півхвилини. Одразу ж
після
цього титрують такий же об’єм розчину аскорбінової кислоти із
мікробюретки
0,001
н
розчином калій йодату
з додаванням у колбочку перед титруванням кількох
кристалів
(близько 5…10
мг) калій йодату
і 2…3
краплель розчину крохмалю, до появи
блакитного
забарвлення. Якщо з’явиться буре або фіолетове забарвлення, слід
приготувати
свіжий
розчин крохмалю. Застосування більшої кількості калій йодату
неприпустиме,
тому
що за високої концентрації цієї солі окислення аскорбінової кислоти
йодом
гальмується.
Розраховують титр барвника (Т) за формулою:
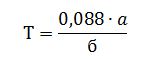
де:
а – кількість точно 0,001 н
розчину калій йодату,
мл;
б
–
кількість розчину барвника, см3.
Приклад розрахунку:
На титрування приготованого розчину аскорбінової кислоти витрачено 2,64 см3 розчину фарби і 3,36 см3 розчину йодату. Отже, 1 см3 розчину фарби еквівалентний (0,088 • 3,36): 2,64 = 0,112 мг аскорбінової кислоти.
Хід аналізу:
- у дві порцелянові ступки вливають по 25 см3 суміші хлоридної
та метафосфорної кислот і зважують їх на технічних терезах з точністю до 0,01
г;
- потім зі спеціально приготованої, грубо подрібненої середньої проби
сировини додають у ці ступки дві наважки по 5…10 г, залежно від вмісту
аскорбінової кислоти в рослинах;
- наважки
з кислотою розтирають
до утворення однорідної маси (грубі
тканини рослин
розтирають
з невеликою, завжди однаковою (приблизно 0,25 г) кількістю скляного
порошку);
- розтерту
масу переносять до мірної колби місткістю 100 см3, зручно
користуватися
колбами
Кольрауша
(рис. 37).
- ступку й товкачик споліскують кілька разів сумішшю кислот, яку
виливають у ту ж мірну колбу;
- цією
сумішшю вміст колби доводять до мітки,
закорковують,
добре перемішують і залишають на п’ять хв.;
- потім частину екстракту (близько 50 см3) фільтрують крізь
сухий подвійний фільтр у суху склянку;
- із отриманого фільтрату беруть піпеткою 10 см3 розчину, наливають у стакан чи колбу і титрують із мікробюретки 0,001 н розчином фарби до рожевого забарвлення, яке не зникає протягом 0,5…1,0 хв.
Розрахунки:
Вміст аскорбінової кислоти (С) у рослинах виражають у мг на 100 г досліджуваної речовини (мг%) та обчислюють за формулою:

де: А – кількість фарби, яку витрачено на титрування екстракту,
см3;
Т
– титр барвника, обчислений за аскорбіновою кислотою,
мг;
В
– об’єм витяжки, отриманої із наважки, см3;
b – кількість фільтрату, витрачена на титрування (зазвичай 10
см3);
а
– наважка досліджуваного матеріалу, г.
Допустима різниця між паралельними визначеннями для картоплі, редиски,
цибул й редьки – 10 %, якщо знайдений відсоток вітаміну С прийняти за 100, а для
інших видів – 5%.
У
рослинних тканинах у деяких кількостях присутні і інші редукуючі речовини, які
відновлюють 2,6-дихлорфеноліндофенол, тому при
необхідності проведення особливо точних аналізів слід взяти до уваги і ці
речовини. Для цього до двох інших порцій по 10…20 см3 досліджуваної
витяжки додають по 0,1 або 0,2 см3 10 % -вого розчину
купрум сульфату і нагрівають на водяній бані 10 хв. при 110 ° С.
Охолоджують і титрують фарбою. У присутності солей міді і при нагріванні
аскорбінова кислота руйнується повністю, без остачі. Отриману поправку
віднімають із даних титрування дослідних розчинів.
Визначення вмісту
вітаміну С у забарвлених витяжках
За аналізу
проб,
витяжки з
яких забарвлені, належить користуватися наступним доповненням до
викладеного
методу:
- беруть наважки,
вилучення та фільтрування здійснюють, як наведено вище;
- потім
з
отриманого
фільтрату беруть піпеткою по 5 см3
і наливають у дві ретельно вимиті й витерті
сухі
пробірки;
- додають у кожну по 3 см3 розчинника – тетрахлорметану
(дихлоретану,
хлороформу або
толуолу);
- обережно збовтують, дають відстоятися протягом півхвилини і титрують із мікробюретки 0,001 н розчином фарби до жовто-рожевого забарвлення.
Техніка
титрування:
- обидві
пробірки беруть в одну руку й повільно по краплях
додають
розчин фарби
з мікробюретки в одну з пробірок, друга є контрольною;
- при
цьому
обидві пробірки струшують одночасно після кожної краплини;
- уважно
слідкують за
зміною
забарвлення розчинника (нижній безбарвний шар) у цій пробірці, порівнюючи
з
контрольною пробіркою на білому
фоні;
- коли з’являється жовто-рожеве забарвлення, титрування
припиняють;
- точно так титрують проби з фільтрату паралельної наважки тієї ж проби й розраховують за формулою, вказаною в основній методиці.
13.1.1.2 Визначення
масової частки аскорбінової кислоти йодометричним методом
Суть методу
Полягає у окисненні
аскорбінової кислоти розчином йоду та визначенні надлишку йоду за допомогою
натрій тіосульфату.
Під
час прямого титрування аскорбінової кислоти розчином йоду відбувається наступна
окисно-відновна реакція:
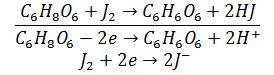
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали: порцелянова ступка, піпетки місткістю 1, 2, 10, 20 см3, товкачик, мірна колба місткістю 100 см3, фільтрувальний папір, конічна колба місткістю 100 см3 та 250 см3, бюретка.
Реактиви:
- Маточний
розчин калій йодату
KJO3:
3,567 г йодноватокислиого
калію (KJO3) у мірній колбі довести до 1000 см3
дистильованою
водою;
- робочий
розчин калій йодату
KJO3:
в мірну колбу на 100 см3 внести 1
см3
маточного розчину KJО3 та довести до позначки дистильованою водою;
- 1%
розчин калій йодиду
(KJ):
в мірну колбу на 100 см3 додати 1
г KJ та довести до позначки дистильованою водою;
- 2%
розчин хлоридної кислоти (НСL):
45,2 см3
НСL довести до 1000 см3 дистильованою водою.
- 1%
розчин крохмалю:
1 г крохмалю заварити в 100 мл води, не доводячи до кипіння, при постійному
перемішуванні.
Підготовка до проведення вимірювань
- відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1. Проби, які надходять у лабораторію повинні бути без пошкоджень і
змін якості продукту при транспортуванні та зберіганні;
- при приготуванні наважки плоди та овочі необхідно подрібнювати ножами і
теркою з нержавіючих матеріалів і як можливо швидше;
Хід аналізу:
- у дві порцелянові ступки вливають по 15…25 см3 2% хлоридної кислоти (НСL) і зважують їх на технічних терезах з точністю до 0,01 г;
- потім зі спеціально приготовленої, грубо подрібненої середньої проби
сировини додають у ці ступки дві наважки по 10…20 г, залежно від вмісту
аскорбінової кислоти в рослинах;
- наважки
з кислотою розтирають
до утворення однорідної маси (грубі
тканини рослин
розтирають
з невеликою, завжди однаковою (приблизно 0,25 г) кількістю скляного
порошку);
- розтерту
масу переносять до мірної колби місткістю 100 см3, зручно
користуватися
колбами
Кольрауша (рис. 37).
- ступку й товкачик споліскують кілька разів сумішшю кислот, яку виливають
у ту ж мірну колбу;
- відстоюють
10 хв. і доводять до позначки (100 см3)
дистильованою водою;
- взбовтують і відфільтровують через фільтр;
Виконують титрування наступним чином:
- дослідний варіант: у
дві колби на 100 см3
вносять
по:
- 15
см3
дистильованої води;
- 1
см3
1 %-вого
розчину калій йодиду
(KJ);
- 2
см3
фільтрату;
- 1
см3
розчину крохмалю у якості індикатору.
- контрольний варіант: у
дві колби на 100 см3
вносять
по:
- 17 см3
дистильованої води;
- 1
см3
1 %-вого
розчину калій йодиду (KJ);
- 1
см3
розчину крохмалю у якості індикатору.
- титрування виконують і дослідних і контрольних варіантів виконують робочим
розчином калій йодату
(KJO3) до появи слабко синього забарвлення.
Розрахунки:
Вміст
аскорбінової кислоти Х,
мг / 100 г
продукту,розраховують за формулою:

де V1
– загальний об'єм витяжки, см3(V1=100 см3);
V2 – об'єм витяжки, який взято для титрування, см3
(V2
=2 см3)
V3 – об'єм калій йодату, який витрачено на титрування дослідного зразка,
см3;
V4 - об'єм калій йодату, який
витрачено на титрування контрольного зразка,
см3;
m – маса наважки, г (m = 10г або 20 г).
Таким чином, підставляючи у формулу 13.2 усі відомі данні отримуємо наступні формули для визначення масової частки аскорбінової кислоти:
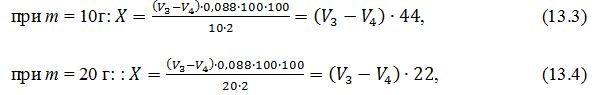
За кінцевий результат випробувань приймають середнє арифметичне результатів двох паралельних визначень, який обчислений до сотих часток відсотка.
13.1.1.3 Визначення
масової частки аскорбінової кислоти фотометричним
методом
Суть методу
Полягає у екстрагуванні вітаміну С метафосфорною кислотою або сумішшю
оцтової і метафосфорної кислот, відновленні 2,6-дихлорфеноліндофенолята натру
аскорбіновою кислотою з наступною екстракцією органічним розчинником
(амілацетатом, бутилацетатом або ксилолом) надлишку 2,6-дихлорфеноліндофенолята
натру і фотометричним визначенням органічного екстракту за довжини хвилі 500
нм.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали:
2 мікробюретки з градуюванням на 0,01 см3, місткістю 2…5
см3; піпетки на 5, 10 і 100 см3; мірні колби на 100, 200,
1000 см3; конічні колби на 100…200 см3; мірні циліндри на
10 і 50 см3; хімічні стакани на 50…100 см3; лійки
діаметром 100…110 мм; порцелянові ступки діаметром 100…120 мм; ножі з
неіржавіючої сталі; скляний порошок (чисте прозоре лабораторне скло, подрібнюють
у ступці і просівають обережно крізь сито з отворами діаметром 1,0…1,5 мм);
центрифуга лабораторна з пробірками та притертими пробками місткістю 25
см3; колориметр фотоелектричний лабораторний або спектрофотометр;
лійки ділильні скляні місткістю 50 см3; прилад для перегонки на
шліфах.
Реактиви: вода дистильована, ацетон, 2,6-дихлорфеноліндофенол натру (фарба Тільманса), розчин масовою концентрацією 0,250 г / дм3; кислота аскорбінова: розчини масовими концентраціями 1,0 і 0,1 г/дм3; кислота нітратна густиною 1,41 г / см3: розчин з об'ємною часткою 25%; кислота метафосфорна: розчини з масовою часткою 3 і 6% (розчин з масовою часткою 3% готують в день випробувань розведенням розчину з масовою часткою 6%, розчин з масовою часткою 6% зберігають в холодильнику протягом 10 днів); кислота хлоридна густиною 1,19 г / см3: розчин з масовою часткою 2%; кислота оцтова льодяна і розчин з масовою часткою 3%; кислота хлоридна, розчин молярної концентрації 0,1 моль/дм3 (готують перед обробкою електродів);
Кислота
етилендиамінтетраоцтова або двонатрієва сіль кислоти: розчин з масовою часткою
5%; калій
йодид:
розчин з масовою часткою 1% в розчині оцтової кислоти з масовою часткою 3%
(готують
перед обробкою електродів); натрій
оцтовокислий плавлений, насичений розчин (готують
наступним чином: 200 г солі розчиняють в 300 см3);
формальдегід,
розчин з масовою часткою 36-40%; аміловий етер (амілацетат); бутиловий етер (бутилацетат); гідрохінон:
напівнасичений розчин в ацетоні; ксилол.
Підготовка до проведення вимірювань
- відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1. Проби, які надходять у лабораторію повинні бути без пошкоджень і
змін якості продукту при транспортуванні та зберіганні;
- при приготуванні наважки плоди та овочі необхідно швидко подрібнювати
ножами і теркою з нержавіючих матеріалів;
-
приготування розчину для екстракції: використовують розчини кислот - хлоридної з масовою часткою 2%,
метафосфорной з масовою часткою 3% або суміші оцтової та метафосфорной кислот,
яку готують наступним чином: 15 г метафосфорной кислоти розчиняють в 250
см3 дистильованої води, додають 40 см3 крижаної оцтової
кислоти , доводять водою до об'єму 500 см3, перемішують і фільтрують
в склянку з притертою пробкою; зберігають в холодильнику не більше 10
днів;
-
приготування стандартних розчинів аскорбінової кислоти: для приготування розчину аскорбінової кислоти концентрацією
1,0 г / дм3 зважують 0,10 г аскорбінової кислоти з
похибкою не більше ± 0,0001 г, розчиняють у розчині для екстрагування в мірній
колбі місткістю 100 см3, доводять до мітки тим же розчином і
перемішують; для приготування розчину концентрації
0,1 г / дм3 вносять піпеткою 10 см3 розчину
аскорбінової кислоти концентрацією 1,0 г / дм3 в мірну
колбу місткістю 100 см3 доводять до мітки розчином для екстрагування
і перемішують (розчини аскорбінової кислоти нестійкі, тому їх готують перед
проведенням випробування);
-
приготування розчину ацетатного буферного, з рН 4: розчиняють 300 г безводного натрій ацетату в 700 см3
дистильованої води, додають 1000 см3 крижаної оцтової кислоти,
перемішують і за допомогою рН- метра
встановлюють рН 4, додаючи, за необхідності, знову
кислоту;
-
приготування розчину 2,6-дихлорфеноліндофенолята натру і визначення його
титру: 0,05 г 2,6-дихлорфеноліндофенолята натру розчиняють приблизно в 150
см3 гарячої води, попередньо прокип'яченої протягом 30 хв.,
охолоджують до кімнатної температури, доводять до об'єму 200 см3 тією
ж охолодженою водою, перемішують і фільтрують в темну склянку, зберігають в
холодильнику не більше 10 днів.
- титр розчину 2,6-дихлорфеноліндофенолята натру встановлюють за
стандартним розчином аскорбінової кислоти концентрацією 1,0 і 0,1
г/дм3 у день проведення випробування. Для цього в дві колби місткістю
50 або 100 см3, в які попередньо додано по 9 см3 води,
вносять піпеткою по 1 см3 розчину аскорбінової кислоти і швидко
титрують розчином 2,6- дихлорфеноліндофенолята натру до світло-рожевого
забарвлення, яке не зникає протягом 15…20 с. Одночасно проводять контрольне
титрування: у колбу місткістю 50 або 100 см3 вносять 1 см3
розчину екстрагування, 9 см3 дистильованої води і титрують розчином
2,6- дихлорфеноліндофенолята натру. Титр розчину
2,6-діхлорфеноліндофенолята натру в грамах аскорбінової кислоти, що
еквівалентний одному кубічному сантиметру розчину 2,6-діхлорфеноліндофенолята
натру, обчислюють за формулою:
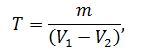
де m - маса аскорбінової кислоти, що міститься в 1 см3 стандартного розчину, г;
V1 - об'єм розчину 2,6-дихлорфеноліндофенолята натру, витрачений на титрування стандартного розчину аскорбінової кислоти, см3;
V2 - об'єм розчину 2,6-дихлорфеноліндофенолята натру, витрачений на
контрольне випробування, см3.
-
приготування напівнасиченого розчину гідрохінону: спочатку готують насичений розчин гідрохінону в ацетоні: 1 г
гідрохінону розчиняють в 10 см3 ацетону і фільтрують.
Напівнасичений розчин гідрохінону готують змішуванням одного об'єму насиченого розчину з таким же
об'ємом ацетону.
-
органічний розчинник не повинен містити окислюючих речовин. Перевірку його чистоти здійснюють наступним чином: до 1 см3
розчину 2,6-дихлорфеноліндофенолята натру спочатку додають розчин аскорбінової
кислоти до знебарвлення, потім додають 10 см3 розчинника, збовтують і
залишають на 10 хв. Якщо органічний шар буде пофарбований, то його слід очистити
перегонкою, збираючи фракцію, яка переганяється відповідно при температурах:
амілацетат - при 149 ° С, бутилацетат - при 126 ° С, ксилол - при
137…141 ° С.
- використаний після випробування органічний розчинник очищують
перегонкою, як зазначено вище. Всі роботи з органічним розчинником слід
проводити у витяжній шафі.
Побудова калібрувального графіка:
- готують п'ять розчинів: в центрифужні пробірки або ділильні лійки
вносять: в першу - 5,0 см3 кислотного розчину для екстрагування, в
інші послідовно по 0,2; 0,4; 0,6; 0,8 см3 розчину
2,6-дихлорфеноліндофенолята натру і доводять розчином для екстрагування до
об'єму 5,0 см3;
- у всі пробірки або ділильні лійки додають по 5 см3 ацетатного
буферного розчину, перемішують;
- потім додають по 10 см3 органічного розчинника;
- пробірки або ділильні лійки закривають пробками і вміст перемішують
протягом 10 с;
- пробірки центрифугують, а лійки залишають у спокої до поділу шарів;
- органічний шар переносять у кювету з відстанню між робочими гранями 10
мм і вимірюють його оптичну густину при довжині хвилі 500 нм;
- в якості контрольного розчину порівняння використовують чистий
розчинник;
- за отриманими даними будують графік залежності оптичної густини
органічного екстракту 2,6-дихлорфеноліндофенолята натру від об'єму розчину 2,6-дихлорфеноліндофенолята
натру в кубічних сантиметрах;
- побудову калібрувального графіка виконують для кожного свіжоприготованого розчину 2,6-діилорфеноліндофенолята натру.
Хід аналізу:
1.Екстрагування:
- для приготування екстракту наважку з середньої проби масою від 5 до 50 г
зважують з похибкою ± 0,01 г;
- для екстрагування вітаміну С з сухих продуктів наважку від 5 до 10 г
розтирають в ступці з невеликими кількостями кислотного розчину для екстракції
або суміші кислот (не менше 1 см3 розчину на 1 г проби) і піску,
переносять в мірну колбу або мірний циліндр місткістю 100 см3,
змиваючи ступку і товкачик невеликими порціями розчину для екстрагування до тих
пір, поки об'єм не досягне позначки; витримують протягом 10 хв, перемішують і
фільтрують.
- для екстрагування вітаміну С з продуктів щільної консистенції наважку
проби від 5 до 50 г гомогенізують не більше 2 хв. з невеликою кількістю
екстрагуючого розчину (не менше 1 см3 розчину на 1 г проби) і
переносять в мірні колбу або циліндр місткістю 100 см3, змиваючи
гомогенізатор невеликими порціями екстрагуючого розчину до тих пір, поки об'єм
не досягне позначки, вміст витримують протягом 10 хв., перемішують і
фільтрують;
- для екстрагування вітаміну С з рідких продуктів наважку проби від 5 до
50 г переносять у мірні колби або циліндр місткістю 100 см3, змиваючи
стінки склянки невеликими порціями екстрагуючого розчину до тих пір, поки об'єм
не досягне позначки; вміст витримують протягом 10 хв., перемішують і
фільтрують;
- при дослідженні продуктів, що містять сульфур (ІІ) оксид
(SO2), наважку проби від 5 до 50 г обробляють в залежності від виду
продукту, як зазначено вище, переносять в мірні колбу або циліндр місткістю 100
см3, додають ацетон в об'ємі, що дорівнює 1/5 частини маси наважки,
перемішують, доводять до позначки екстрагуючим розчином; вміст витримують 10
хв., знову перемішують і фільтрують;
- при дослідженні продуктів, фасованих в металеву тару, наважки проби від
5 до 30 г обробляють в залежності від виду продукту, як зазначено вище,
переносять у мірну колбу або циліндр місткістю 100 см3 за допомогою
екстрагуючого розчину, доводять до об'єму 50 см3 і перемішують; через
10 хв. додають 10 см3 насиченого натрій ацетату або 30 см3
ацетатного буферного розчину, додають 10 см3 розчину
етилендіамінтетраоцтової кислоти або її солі, перемішують, доводять до позначки
екстрагуючим розчином, знову перемішують і фільтрують.
2.Визначення:
- у центрифужну пробірку або ділильну лійку вносять піпеткою від 1 до
5 см3 отриманого екстракту випробуваної проби;
- додають розчин для екстрагування до об'єму 5 см3, такий же
об'єм ацетатного буферного розчину і розчин 2,6-дихлорфеноліндофенолята натру в
об'ємі не більше 2 см3;
- перемішують і додають 10 см3 органічного розчинника;
- пробірки центрифугують, а лійки залишають у спокої до поділу шарів;
- органічний шар переносять у кювету з відстанню між робочими гранями 10
мм і вимірюють його оптичну густину при довжині хвилі 500 нм;
- в якості контрольного розчину порівняння використовують чистий розчинник;
* При отриманні мутного органічного екстракту перед вимірюванням оптичної густини екстракт фільтрують через фільтрувальний папір.
Одночасно проводять контрольне титрування на вміст у продукті редукуючих речовин:
- в центрифужну пробірку або ділильну лійку вносять досліджуваної проби 1
до 5 см3, додають розчин для екстрагування до об'єму 5
см3, вносять такий же об'єм ацетатного буферного
розчину;
- додають розчин формальдегіду в об'ємі, рівному половині об'єму буферного
розчину, перемішують і витримують протягом 10 хв.
- після цього додають розчин 2,6-дихлорфеноліндофенолята натру, знову
перемішують і додають 10 см3 органічного розчинника;
- пробірки центрифугують, а лійки залишають у спокої до поділу шарів;
- органічний шар переносять у кювету з відстанню між робочими гранями 10
мм і вимірюють його оптичну густину при довжині хвилі 500 нм;
- в якості контрольного розчину порівняння використовують чистий
розчинник;
При вмісті в продукті розчинних в органічному розчиннику барвних речовин
їх вплив визначають наступним чином:
- після проведених визначень за пунктом 2 в кювету з органічним екстрактом
додають дві краплі напівнасиченого розчину гідрохінону,
- перемішують паличкою, витримують 30 с,
- знову вимірюють оптичну густину;
- отримане значення оптичної густини віднімають від початкового значення
оптичної густини органічного екстракту.
Розрахунки:
Масову частку аскорбінової кислоти (Х) у відсотках обчислюють за формулою:

де V1- об'єм розчину 2,6-дихлорфеноліндофенолята натру, витрачений на
проведення випробування, см3;
V2- об'єм надлишку розчину
2,6-дихлорфеноліндофенолята натру, знайдений за калібрувальним графіком,
см3;
V3 - об'єм розчину 2,6-дихлорфеноліндофенолята натру, витрачений на
контрольне титрування, см3;
T- титр розчину 2,6-дихлорфеноліндофенолята натру,
г/см3;
V4- об'єм екстракту, отриманий під час добування вітаміну С з наважки
продукту, см3;
V5 - об'єм екстракту, який використовували для випробування,
см3;
m - маса наважки продукту, г.
За остаточний результат випробування приймають середнє арифметичне
результатів двох паралельних визначень. Обчислення проводять до чотирьох
значущих цифр після коми, результат округлюють до трьох значущих цифр і
висловлюють у вигляді множення числа на 10-3.
Розбіжність між двома паралельними визначеннями не повинна перевищувати 3% від середнього арифметичного значення з вірогідністю 0,95.
13.1.1.4 Кількісне
визначення аскорбінової, дегідроаскорбінової та дикетогулонової кислот у
рослинних тканинах (за
Г. Н. Чупахіною)
Аскорбінова кислота (АК) – сполука, яка легко окиснюється до дегідроаскорбінової кислоти (ДАК), лактонове кільце якої легко гідролізується з утворенням кислоти з відкритим ланцюгом - дикетогулонової кислоти (ДКГК).
Суть методу
Кількісного визначення даних кислот грунтується на взаємодії
2,4-динітрофенілгідразину з ДАК і ДКГК із утворенням у сульфатній кислоті
відповідних азазонів. Азазони ДАК і ДКГК дають червоне забарвлення, яке
використовується для фотометричного визначення. Для знаходження суми всіх кислот
АК окиснюють розчином 2,6-дихлорфеноліндофенола
до ДАК і вміст відновленої форми АК визначають за різницею. При роздільному
визначенні ДАК і ДКГК суміш піддають дії відновників, при цьому відновлюватися в
АК може тільки ДАК. В. В. Соколовський, Л. В. Лебедєва,
Т. Б. Ліелуп у якості відновника запропонували використовувати унітіол
(димеркаптопропансульфонат натру), що значно спростило процедуру відновлення.
При рН 7 унітіол протягом 10 хв. відновлює ДАК в АК.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали:
порцелянова ступка з товкачиком, гомогенізатор, мірна колба на 25
см3, центрифуга, центрифужні пробірки, градуйовані пробірки з
притертими пробками, холодильник побутовий, термостат (або водяна баня Т=100°С),
льодяна баня, спектрофотометр або фотоелектроколориметр, бюретки, піпетки,
скляні палочки.
Реактиви:
- 2 % - вий розчин 2,4-динітрофенілгідразину в 9 н.
сульфатній кислоті, який містить 0,25% тіосечовини: для приготування 100 см3 реактиву необхідно розрахувати, яку
кількість концентрованої сульфатної кислоти необхідно взяти з урахуванням її
концентрації і густини, щоб кінцевий розчин був 9 н. Наприклад, вихідний розчин
концентрованої сульфатної кислоти 95,6% і густина його 1,830. В цьому випадку
необхідно взяти 25,2 см3 кислоти, в кислоті при нагріванні або
струшуванні розчинити 2 г 2,4-динітрофенілгідразина, а потім 0,25 г тіосечовини;
отриманий розчин змішати з необхідним об'ємом води, наливаючи кислотний розчин в
воду так, щоб кінцевий об'єм дорівнював 100 см3; розчин зберігають у
холодильнику не більше одного місяця.
- 5 % - ва метафосфорна
кислота (зберігати в холодильнику не більше двох тижнів);
- 85 % - вий розчин сульфатної
кислоти: в 100 см3 дистиляту влити 900 см3 концентрованої
сульфатної кислоти;
- 2•10-3 М унітіол: 0,84 см3 5 % - вого розчину ампульованого
препарату в 100 см3 фосфатного буфера 0,2 М, рН = 7 (розчин зберігати
не більше доби);
- 0,001 н. розчин 2,6-дихлорфеноліндофенола (фарба Тильманса), зберігати в темряві не більше одного тижня.
Підготовка до проведення вимірювань
- відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
- проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні;
- при приготуванні наважки плоди та овочі необхідно швидко подрібнювати ножами і теркою з нержавіючих матеріалів.
Хід аналізу:
- наважку рослинного матеріалу (близько 0,5 г) залити в порцеляновій
ступці 10 см3 5 % - вої метафосфорної кислоти,
розтерти;
- гомогенат кількісно перенести у мірну колбу на 25
см3;
- об'єм довести до позначки метафосфорною кислотою;
- після центрифугування протягом 20 хв. за 3000 об./хв. відокремити
осад;
- у дві градуйовані пробірки з притертими пробками налити по 1,5
см3 отриманої витяжки;
- в одну з них додати по краплях 0,001 н розчин 2,6-дихлорфеноліндофенола
до появи слабко-рожевого забарвлення, стійкого протягом 30
секунд;
- у третю пробірку налити 1,5 см3 витяжки, приготовленої на
2•10-3 М розчині унітіолу, яка готується наступним чином: наважку
матеріалу (близько 0,5 г) розтерти з розчином унітіолу, перенести в мірну колбу
на 25 см3, об'єм довести до позначки розчином унітіолу, що
приготовлений на фосфатному буфері; вміст колби струсити, колбу помістити в
холодильник на 10 хв. - це час, необхідний для відновлення ДАК в АК.
Центрифугуванням відокремити осад. Білки, що знаходяться у витяжці, осадити
метафосфорною кислотою: до 16 см3 центрифугата додати 4
см3 5 % - вої метафосфорної кислоти, білки які випали в
осад видалити центрифугуванням (15 хв., 3000 об./хв.).
- у всі три пробірки додати по 0,5 см3
2,4-динітрофенілгідразину і довести об'єм до 2,5 см3 дистильованою
водою;
- пробірки помістити в термостат на 20 хв. за температури
100 ° С;
- після закінчення зазначеного часу пробірки перенести в крижану
баню;
- в кожну з них додати по 2,5 см3 (трьома порціями) сульфатної
кислоти;
- через годину забарвлені розчини фотометрують за довжини хвилі 520 нм в
кюветі на 10 мм в порівнянні з контрольним розчином, який готується і
обробляється як дослідні, тільки замість витяжки використовується розчин
5 %- вої метафосфорної кислоти;
- за калібрувальним графіком визначають, якій концентрації кислоти відповідає дана оптична густина.
Розрахунки:
Вміст кислоти Х на 1 г
наважки визначається за формулою:

де С - концентрація розчину кислоти, яка відповідає даній оптичній густині, мкг / мл;
n - наважка рослинного матеріалу, г;
25 - загальний об'єм досліджуваного розчину, см3;
1,5 - об'єм розчину, взятий для аналізу, см3.
Кількісний вміст кислот далі розраховується наступним
чином:
- у пробірці з екстрактом, отриманим з унітіолом, визначається тільки
ДКГК, так як унітіол відновлює ДАК в АК, яка з 2,4-динітрофенілгідразином
не визначається;
- у цю величину необхідно ввести поправку на розведення за рахунок
використання метафосфорної кислоти для осадження білків: отриманий результат
потрібно збільшити на 20%;
- у пробірці з кислотним екстрактом визначається ДАК і ДКГК, тому,
віднявши від даної суми кількість ДКГК, знайдемо величину
ДАК;
- у пробірці з кислотним екстрактом, де АК була окислена
2,6-дихлорфеноліндофенолом до ДАК, визначається сума трьох кислот (АК, ДАК і
ДКГК);
- знаючи значення ДАК і ДКГК, знаходимо за різницею величину
АК.
За остаточний результат випробування приймають середнє арифметичне
результатів двох паралельних визначень. Обчислення проводять до чотирьох
значущих цифр після коми, результат округлюють до трьох значущих цифр і
висловлюють у вигляді множення числа на 10-3.
Розбіжність між двома паралельними визначеннями не повинна перевищувати
3% від середнього арифметичного значення з вірогідністю 0,95.
Побудова калібрувального графіка:
Для побудови калібрувального графіка готують три розчини аскорбінової
кислоти в 5 % - вій метафосфорній кислоті:
- 1-й розчин - 100 мг АК в 250 см3 метафосфорної
кислоти;
- 2-й розчин – до 0,5 см3 1-го розчину додають 9,5
см3 метафосфорної кислоти;
- 3-й розчин – до 0,5 см3 2-го розчину додають 4,5
см3 метафосфорної кислоти.
Певні об'єми 1-го і 2-го розчинів (1 см3, 075 см3,
0,5 см3, 025 см3 - 2-го розчину і 1 см3, 0,5
см3 - 3-го розчину), що містять від 20 до 2 мкг АК, доводяться
метафосфорною кислотою до 1,5 см3. В якості контролю береться 1,5
см3 кислоти.
У всі пробірки по краплях додається 0,001 н розчин 2,6-дихлорфеноліндофенолу
до появи слабко-рожевого забарвлення, стійкого протягом 30 секунд (АК перейде в
ДАК).
Потім вноситься по 0,5 см3 розчину 2,4-динітрофенілгідразину
і об'єм доводиться до 2,5 см3 дистильованою водою. Далі проби
обробляються наступнним чином:
- пробірки помістити в термостат на 20 хв. за температури
100 ° С;
- після закінчення зазначеного часу пробірки перенести в крижану
баню;
- в кожну з них додати по 2,5 см3 (трьома порціями) сульфатної
кислоти;
- через годину забарвлені розчини фотометрують за довжини хвилі 520 нм в
кюветі на 10 мм в порівнянні з контрольним розчином, який готується і
обробляється як дослідні, тільки замість витяжки використовується розчин
5 %- вої метафосфорної кислоти;
- за показниками оптичної густини для розчинів ДАК різної концентрації, отриманих за допомогою фотоелектроколориметра, будується калібрувальний графік.
13.1.2 Визначення вмісту
рутина
Термін «вітамін Р» є комплексним поняттям. Цим терміном об'єднується
ціла група речовин, які мають подібну біологічну дію – зміцнювати стінки
кровоносних судин і разом із вітаміном С запобігати захворюванням.
Вітамін Р зазвичай міститься в тих же рослинах, в яких зустрічається і
аскорбінова кислота, вони вважаються синергістами.
В даний час з рослин виділено значну кількість речовин, які володіють Р-вітамінною активністю. Серед них найбільше значення мають: рутин, що отримується з листя гречки (4%); гесперидин (цитрин), що виділяється з шкірки цитрусових; катехіни і їх галові етери, які отримують із чайного листа (20%). Середній вміст рутину становить(в мг %): шипшина - 1000, чорноплідна горобина (листя) - 4000, чорна смородина-1000, лимони - 500, вишня - 2000, морква - 100, агрус - 500, петрушка - 150, квітки софори японської - 20, плоди горобини чорноплідної - 20, лист мати-і-мачухи - 0,5. Потреба людини у вітаміні Р орієнтовно - 50 мг за добу.
Суть методу
Полягає у вилученні рутину з рослинного матеріалу спиртом і визначенні в
очищеному спиртовому екстракті його вміст колориметрично за інтенсивністю
забарвлення з алюміній (ІІІ) хлоридом алюмінієм у присутності надлишку натрій
ацетату.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: центрифуга, спектрофотометр або фотоелектроколориметр, ступки
порцелянові, скляні фільтри, лійки Бюхнера, колби Вюрца (рис. 11), чашки
порцелянові, колби мірні на 100 і 50 см3, пробірки, лійки, фільтри,
стакани.
Реактиви: рутин кристалічний, етиловий спирт, 2 % - вий розчин АlС13, 8 % - вий CH3COONa.
Підготовка до проведення вимірювань
- відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1. Проби, які надходять у лабораторію повинні бути без пошкоджень і
змін якості продукту при транспортуванні та зберіганні;
- при приготуванні наважки плоди та овочі необхідно подрібнювати ножами і
теркою з нержавіючих матеріалів і як можливо швидше;
Побудова калібрувального графіка:
- готують стандартний розчин рутину шляхом розчинення 20 мг чистого рутину
в 100 см3 80 % - вого етилового спирту;
- для кращого розчинення рутину розчин можна злегка підігріти;
- потім в широкі пробірки вносять такі кількості стандартного розчину
рутина (в см3): 1,0; 1,5; 2,0; 2,5; 3,0; 4,0; 4,5; 5,0;
- об'єм кожного розчину доводять до 5 см3 80 % -вим
етиловим спиртом;
- в отриманому об'ємі кожної пробірки відповідно міститься 0,2; 0,4; 0,5;
0,6; 0,7; 0,8; 0,9 і 1,0 мг рутину;
- потім у кожну пробірку додають по 5 см3
2 % - го розчину А1С13 і по 15 см3
8 % - го розчину CH3COONa, перемішують і залишають
стояти в темному місці 2 години;
- для побудови калібрувального графіка на осі абсцис відкладають міліграми рутину, а на осі ординат - оптичну густину забарвлених розчинів.
Хід аналізу:
- наважку рослинного матеріалу масою 2…10 г (залежно від передбачуваного
вмісту рутину) переносять у порцелянові ступки;
- ретельно розтирають з невеликою кількістю кварцового піску в присутності
спирту;
- розтерту масу переносять на скляний фільтр або на лійку Бюхнера;
- ступку ретельно споліскують спиртом, залишки рослинної маси переносять
на фільтр;
- рослинний матеріал на фільтрі або лійці Бюхнера багаторазово екстрагують
спиртом до повного знебарвлення залишку і стікання екстракту;
- екстракт фільтрують за допомогою водоструминного насосу;
*Рутин можна також
екстрагувати з рослинних зразків шляхом тривалого настоювання зі спиртом із
наступним відділенням залишку центрифугуванням.
- екстракт переносять у мірну колбу місткістю 100 см3 і
доводять спиртом до позначки;
- з цього обєму беруть 25 см3 для аналізу і переносять в
порцелянову чашку або колбу Вюрца;
- спирт видаляють з порцелянової чашки випаровуванням на водяній бані, а з
колби Вюрца – відгонкою;
- після видалення спирту до осаду в чашці або в колбі Вюрца додають малими
порціями етер для розчинення пігментів (хлорофілу, каротиноїдів та інших
етеророзчинних речовин);
- за допомогою скляної палички етерні витяжки і осад з чашки або колби
переносять на фільтр;
- промивають осад на фільтрі етером і етерні витяжки відкидають;
- рутин на фільтрі розчиняють гарячим етиловим
спиртом;
- загальний об'єм екстракту доводять у мірній колбі 80 % - вим етанолом до об'єму 50 см3;
**Цей розчин використовують для колориметричного визначення вмісту рутина. Якщо під час екстрагування рутину з рослинних зразків отримують безбарвні екстракти, то їх можна відразу ж використовувати для аналізу, минаючи обробку зразків етером.
В іншому випадку готують розчин для колориметрування наступним чином:
- в широку пробірку або в невелику колбу наливають 5 см3
спиртового розчину рутину,
- потім додають 5 см3 2 % - вого розчину
хлористого алюмінію і 15 см3 8 %- вого розчину натрій
ацетату;
- вміст пробірки або колби ретельно перемішують і залишають стояти на 2
години в темному місці (за цей час розчин набуває стійкого жовтого
забарвлення);
- одночасно готують контрольний розчин: готують таким же чином, як і
дослідний розчин, тільки замість алюміній (ІІІ) хлориду беруть 5 см3
води, 15 см3 8 % - го розчину натрій ацетату,
перемішують і залишають стояти 2 години;
- для колориметрування використовують кювету з товщиною шару 10
мм;
- колориметрування проводять проти контролю при синьому світлофільтрі (420
нм);
- вміст рутину обчислюють за калібрувальним графіком, побудованому раніше.
Розрахунки:
Вміст рутину (Х), виражений у міліграмах на 100 г рослинного матеріалу (мг %), визначають за формулою:

де C
– вміст рутина у 5 см3 екстракту, визначений за калібрувальним
графіком, мг,
V1
– загальний об'єм спиртового екстракту, см3;
V2
– об'єм спиртового розчину рутину після обробки етером,
см3;
V3
–
об'єм екстракту, який беруть для аналізу, см3;
V4
– об'єм екстракту, взятий для колориметрування,
см3;
m
– маса наважки, г.
Приклад обчислення результатів. Наважка рослинного матеріалу - 10 г. Загальний об'єм спиртового екстракту - 100 см3. Для аналізу беруть 25 см3 екстракту. Після обробки етером об'єм спиртового розчину рутину становить 50 см3. Для колориметрування взято 5 см3 екстракту. За калібрувальним графіком у 5 см3 екстракту міститься 0,6 мг рутину. Вміст рутину (Х), виражений у міліграмах на 100 г рослинного матеріалу (мг %), становитиме:
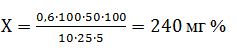
За остаточний результат випробування приймають середнє арифметичне
результатів двох паралельних визначень. Обчислення проводять до чотирьох
значущих цифр після коми, результат округлюють до трьох значущих цифр і
висловлюють у вигляді множення числа на 10-3.
Розбіжність між двома паралельними визначеннями не повинна перевищувати
3% від середнього арифметичного значення з вірогідністю
0,95.
13.1.3 Визначення загального вмісту токоферолів колориметричним
методом
Вітамін Е являє собою суміш різних форм токоферолу, які володіють
біологічною або антиокислювальною активністю. Найбільшою фізіологічною дією
характеризується α – токоферол (С29Н50О2), а
антиоксидантною активністю – δ-токотриенолу
(С27Н40О2). Всі форми токоферолу легко
окиснюються, і тому важливо знати вміст активної, постійної частини вітаміну в
пробі. Вміст вітаміну Е у цілих плодах обліпихи – 8…16 мг %, в олії з
обліпихи - до 200 мг%, в кукурудзяній олії - до 500, в олії з пшеничних зародків
- 300 мг%.
Суть методу
Грунтується на відновленні токоферолами заліза (ІІІ) хлориду і утворенні
двовалентним залізом з дипіридином або ортофенантроліном продукту
рубіново-червоного кольору.
Омилення проводять в струмі нітрогену в колбі місткістю 50
см3 з повітряним холодильником при нагріванні на водяній
бані.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали: апарат Сокслета, фотоелектроколориметр, колби мірні на 25 см3; колби конічні, колби круглодонні, піпетки, бюретки, фільтри скляні №3, ступка порцелянова з товкачиком, водяна баня, лійка, палички склянні, ексикатор, абсорбційна колонка, терези аналітичні.
Реактиви: пірогалол, метанол, етанол, вода дистильована, 10 % - вий та 60 % - вий спиртові розчини КОН, диетиловий етер, фенолфталеїн, безводний натрій сульфат, бензол, 0,5 % - вий спиртовий розчин ортофенантраліна, адсорбенти – діатоміт або силікагель.
- розчин заліза (ІІІ) хлориду: 0,2 % - вий розчин FeCl3•H2O в абсолютному етиловому спирті;
- розчин α1α'-дипіридина: 0,5 % -вий розчин α1α'-дипіридина в абсолютному етиловому спирті.
Підготовка до проведення вимірювань
- відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
- проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні;
Побудова калібрувального графіка:
- готують стандартний розчин токоферола: на анлітичних терезах зважують 50
мг α-токоферолу,
- розчиняють його в бензолі в мірній колбі на 250
см3;
- із отриманого стандартного розчину в мірні колби на 25 см3
вносять по 0,25; 0,50; 0,75; 1; 1,25; 1,50; 1,75; 2
см3;
- додають α1α'-дипіридина або ортофенантраліна,
FeCl3 і об'єм доводять до позначки;
- через 10 хв. колориметрують;
- при побудові калібрувального графіку на осі абсцис відкладають концентрацію розчину ( в мг або в мкг), а на осі ординат – екстинцію розчинів.
Хід аналізу:
- наважку 1…2 г рослинного матеріалу розтирають у ступці зі склянним
піском у присутності етанолу або суміші етанолу та гексану
(1:4);
- розтерту масу промивають на скляному фільтрі №3 невеликими порціями
спирту (20…30 см3) до безбарвного фільтрату;
- потім промивають етером: беруть приблизно половинний об'єм взятого
спирту;
- загальний об'єм етерно-спиртової суміші становить 30…50
см3;
- до етерно-спиртової суміші додають 60 % -вий розчин КОН у
кількості до кінцевої концентрації 10 % (на 30…50 см3 приблизно 6…10
см3 60 % -вого розчин КОН)
- омилюють на киплячій водяній бані протягом години у присутності 0,3 г
пірогалолу;
- після омилення суміш кількісно переносять у ділильну
лійку,
- розчиняють такою ж самою кількістю води та обережно
перемішують;
- у нижній, водний шар переходить сіль хлорофілу, а у верхній, етерний –
токофероли і каратиноїди;
- етерний шар збирають;
- водно-спиртовий шар декілька разів промивають етером та
відкидають;
- етерні екстракти поєднують, сушать у ексикаторі натрій
сульфатом;
- переносять у круглодонну колбу і розчинник
відгоняють;
- залишок розчиняють у бензолі і кількісно переносять у мірну колбу на 25 см3;
Адсорбція і колориметрування:
- із отриманого розчину беруть 5…10 см3 та вносять на колонку з
діатомітом або силікагелем, пропускаючи розчин при слабкому розрідженні (висота
шару 3,5 см, диаметр 1,5 см);
- змивають токофероли з адсорбенту 12…18 см3
бензолу;
- елюати кількісно збирають у мірній колбі на 25 см3 та
доводять об'єм до позначки бензолом;*
- для барвної реакції беруть 5…10 см3 отриманого
розчину;
- вносять у мірну колбу на 25 см3, приливають 10…15
см3 бензола і 1 см3 0,5 % -вого розчину
α1α'-дипіридина або 0,5 % - вого розчину
ортофенантраліна;
- потім по краплям, при постійному перемішуванні приливають 1
см3 заліза (ІІІ) хлориду;
- об'єм доводять до позначки бензолом, перемішують та залишають у темному
місці на 10 хв.;
- одночасно ставлять контроль, приливаючи такіж самі кількості реактивів у
мірну колбу на 25 см3;
- розчини колориметрують на фотоелектроколориметрі за довжини хвилі 490
нм, встановлюючи нульову позначку за бензолом;
- використовують кювету з робочою довжиною 5 мм;
- від значення оптичної густини досліджуваного розчину віднімають показник контролю.
Розрахунки:
Вміст токоферолів за масою проби (Х),
обчислюють за
формулою:

де a – вміст
токоферолів, яке знайдено за калібрувальним графіком;
V – об'єм початкового екстракту,
см3, (25 см3);
V1 - кількість розчину для
адсорбції на колонку, см3;
V2 – об'єм елюату після
пропускання через колонку з діатомітом або силікагелем (25
см3).
V3
– кількість розчину, який взято для проведення кольрової
реакції;
m – маса наважки, г.
* Замість бензолу зручніше використовувати абсолютний етанол. При цьому: під час адсорбції отримані бензольні елюати збирають, та перегоняють у потоці нітрогену.
До залишку додають 10 см3 абсолютного спирту, нагрівають
15…20 с на водяній бані при 60 °С та переносять у мірну колбу на 25
см3. Під час барвної реакції доведення до позначки виконують
абсолютним спиртом. Колориметрують проти спирту. Для побудови калібрувального
графіку та колориметрування також використовують спирт.
За остаточний результат випробування приймають середнє арифметичне
значення двох паралельних визначень, розрахованих до четвертого і округлених до
третього десяткового знака, розбіжність між якими при Р = 0,95 не
повинна перевищувати 13% по відношенню до середнього арифметичного значення.
13.1.4 Визначення вмісту
вітаміну РР (ніацину)
Метод поширюється на продукти переробки плодів і овочів.
Суть методу
Полягає у звільненні вітаміну РР шляхом гідролізу, очищення гідролізату
від заважаючих визначенню речовин, кількісному отриманні пофарбованого
глутамінового альдегіду, інтенсивність забарвлення якого вимірюють
фотометрично.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: колориметр фотоелектричний і кварцовими кюветами робочої довжиною 30 мм;
або спектрофотометр, забезпечений кварцовими кюветами з робочою довжиною 10
мм; терези лабораторні з найбільшою
межею зважування до 200 г; терези лабораторні з найбільшою межею зважування до 1
кг; електроплитка побутова; баня водяна; баня льодяна; бюретки; колби
круглодонні, колби конічні; циліндри на 50, 100 см3; склянки на 1000,
2000 см3; лійки Бюхнера № 2, 3 або 4; лійки лабораторні; термометр
рідинний скляний з межею похибки 2 ° С; піпетки на 1, 2, 5, 10
см3; ексикатор, палички з хіміко-лабораторного скла, пробірки, ступка
порцелянова з товкачиком.
Реактиви: вітамін РР (ніацин); кислота сульфатна, х.ч.: розчини с(H2SO4)= 0,05 моль/дм3, с(H2SO4)= 1,0 моль / дм3 і с(H2SO4) = 2,5 моль / дм3; кислота хлоридна, х.ч.,
розчини с(HCl) = 0,5 моль / дм3 і с(HCl)= 5 моль / дм3; натру гідроокис, х.ч., розчини
с(NaOH)= 4,0 моль / дм3, с(NaOH) = 10 моль / дм3; бром, х.ч.; калій роданистий, х.ч., або амоній
роданистий, х.ч., розчини масової концентрації 100 г / дм3
і 10 г/дм3; кальцій вуглекислий, х.ч.; кальцій гідроксид, ч; метол,
перекристалізований, розчин масовою концентрацією 80 г / дм у хлорній
кислоті с(HCl) = 0,5 моль / дм3; спирт етиловий ректифікований технічний;
цинк сульфат, х.ч., розчин масовою концентрацією 800
г / дм3; фенолфталеїн, розчин масової концентрації 10
г / дм3; вугілля активоване, вода дистильована, папір
фільтрувальний лабораторний, фільтри знезолені.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1. Проби, які надходять
у лабораторію повинні бути без пошкоджень і змін якості продукту при
транспортуванні та зберіганні;
Приготування основного стандартного розчину вітаміну РР
концентрації 0,1 мг/см3: наважку вітаміну РР масою 0,050 г поміщають в мірну колбу місткістю 500
см3, додають 300 см3 води і 5 см3 розчину
сульфатної кислоти с(H2SO4) = 2,5 моль / дм3; після розчинення вітаміну РР
доводять обсяг до мітки водою, ретельно перемішують і переносять у склянку з
темного скла з притертою пробкою. Зберігають розчин в холодильнику не більше 3
міс.
Приготування робочого стандартного розчину вітаміну РР
концентрації 2 мкг/ см3: в день проведення аналізу 2 см3 основного стандартного
розчину вітаміну РР піпеткою вносять в мірну колбу місткістю 100 см3,
об'єм доводять водою до мітки і ретельно перемішують.
Приготування бромної води: за 5 днів до проведення аналізу в склянку темного скла з притертою
пробкою вносять 100 см3 води і (у витяжній шафі) 5…6 см3
брому, енергійно струшують. Склянку з бромної водою зберігають у витяжній шафі в
захищеному від світла ексикаторі.
Приготування роданбромідного розчину: роданбромідний розчин готують у витяжній шафі безпосередньо перед
застосуванням. До охолодженої на льоду бромной води, взятої в об'ємі,
необхідному для аналізу, додають по краплях охолоджений розчин роданистого калію
або амонію масовою концентрацією спочатку 100 г / дм3,
потім 10 г / дм3 до повного знебарвлення брому. Після
цього поступово, невеликими порціями вносять вуглекислий кальцій до припинення
виділення бульбашок газу і утворення осаду. Розчин фільтрують у склянку з
темного скла з притертою пробкою і залишають на крижаній
бані.
Перекристалізація метола
У склянці місткістю 1000 см3 нагрівають до кипіння 500 см3 розчину сульфатної кислоти с(H2SO4) = 0,05 моль / дм3, додають 100 г метола і продовжують нагрівати суміш до початку кипіння. Якщо розчин сильно забарвлений, то до нього додають 10 г активованого вугілля, перемішують і відразу ж фільтрують через лійку для гарячого фільтрування або лійку Бюхнера, попередньо нагріту киплячою водою, в хімічний стакан місткістю 2000 см3. До фільтрату додають 700 см3 етилового спирту, перемішують, стакан поміщають у крижану баню і залишають в темряві на 4…5 годин. Випавший осад фільтрують через лійку Бюхнера, кристали на фільтрі промивають невеликими (по 30…40 см3) кількостями охолодженого спирту і висушують на повітрі в темряві. Перекристалізований метол зберігають у склянці з темного скла з притертою пробкою в захищеному від світла місці.
Приготування розчину метола
Безпосередньо перед застосуванням в мірній колбі місткістю 100 см3 розчиняють 8,0 г перекристалізованого метола в розчині хлоридної кислоти с(HCl)= 0,5 моль / дм3. Доводять об'єм до мітки розчином хлоридної кислоти.
Хід аналізу:
1.Гідроліз
Залежно від виду досліджуваного продукту для звільнення зв'язаних форм
вітаміну РР використовують різні способи гідролізу.
При аналізі продуктів переробки плодів і овочів без і з додаванням круп,
молока і сиру застосовують лужний гідроліз:
- наважку проби масою 1,0…10,0 г розтирають у фарфоровій ступці з 1,5 г
кальцій гідроксиду;
- потім кількісно переносять вміст ступки в конічну колбу місткістю 100
см3, змиваючи 50-60 см3 води невеликими
порціями;
- колбу з пробою нагрівають протягом 90 хв. на киплячій водяній бані,
попередньо закривши шийку колби маленької лійкою або спеціальною скляною
пробкою-вкладишем, періодично струшуючи;
- після нагрівання колбу охолоджують до кімнатної температури;
- потім об'єм гідролізату доводять водою до 75 см3,
перемішують, охолоджують 2 години на крижаній бані або залишають на ніч в
холодильнику;
- охолоджений гідролізат фільтрують або центрифугують;
- 25 см3 фільтрату поміщають в циліндр місткістю 50 см3, додають 1…2 краплі розчину фенолфталеїну і по краплях розчин сульфатної кислоти с(H2SO4) = 2,5 моль / дм до знебарвлення. Далі проводять обробку, як зазначено в п.2.
При аналізі продуктів переробки овочів з додаванням м'яса, риби,
яєць застосовують кислотний гідроліз:
- наважку проби масою 1,0…10,0 г кількісно переносять в конічну колбу
місткістю 100 см3, змиваючи 50…60 см3 розчину сульфатної
кислоти с(H2SO4) = 1 моль / дм3 невеликими порціями.
- колбу з пробою нагрівають протягом 90 хв. на киплячій водяній бані,
попередньо закривши шийку колби маленької лійкою або спеціальною скляною
пробкою-вкладишем, періодично струшуючи;
- потім колбу охолоджують до кімнатної температури;
- об'єм гідролізату доводять водою до 75 см3, ретельно
перемішують і фільтрують через складчастий фільтр (перші 5…6 см3
фільтрату відкидають);
- 25 см3 фільтрату поміщають в циліндр місткістю 50
см3, додають 1…2 краплі фенолфталеїну і нейтралізують розчином їдкого
натру с(NaOH) = 10 моль / дм3 до слабо-рожевого забарвлення
і охолоджують;
- надлишок лугу усувають додаванням 1…2 крапель розчину сульфатної кислоти
с(H2SO4) = 2,5 моль / дм3.
-далі проводять обробку, як зазначено в п.2.
Якщо фільтрати інтенсивно пофарбовані, то рН розчину доводять до 8 за
універсальним індикаторним папером розчинами кислоти або лугу тих же
концентрацій, які застосовувалися при гідролізі.
2.Очищення фільтрату:
- у колбу з нейтральним фільтратом вносять 2 см3 розчину цинк
сульфату;
- потім вносять розчин натрій гідроксиду с(NaOH) = 4 моль / дм3 по краплях до слабо-рожевого
забарвлення;
- ретельно перемішують, видаляють рожеве забарвлення додаванням 1…2
крапель розчину сульфатної кислоти
с(H2SO4) = 2,5 моль / дм3;
- витримують протягом 10 хв., потім додають 1…2 краплі етилового спирту
для усунення піни;
- об'єм доводять водою до позначки, перемішують і фільтрують через складчастий фільтр.
3. Проведення кольорової реакції:
- для проведення кольорової реакції використовують 8 пробірок або колб з
притертими пробками;
- у три пробірки піпеткою вносять по 5 см3 робочого
стандартного розчину вітаміну РР,
- у 4 пробірки вносять піпеткою по 5 см3 отриманого
фільтрату,
- в контрольну пробірку - 5 см3 води замість
фільтрату;
- всі пробірки закривають пробками і нагрівають на водяній бані при
температурі 48…52 °С протягом 5…10 хв.;
- потім в пробірки зі стандартним розчином вітаміну, в пробірку з водою і
в дві пробірки з досліджуваним фільтратом додають з бюретки у витяжній шафі по 2
см3 роданбромідного розчину;
- всі пробірки закривають пробками, струшують і залишають на водяній бані
при температурі (50 ± 2) °С 10 хв.;
- через 10 хв. всі пробірки охолоджують водою до кімнатної температури,
залишають на 10 хв в темному місці;
- потім в кожну з них додають по 3 см3 розчину метола,
струшують і залишають на 1 годину в темному місці;
- після цього вимірюють оптичну густину розчинів на фотоелектроколориметри
(400…425 нм) або спектрофотометрі (420 нм). Контрольним розчином служить
дистильована вода;
- якщо розчини каламутні, то перед вимірюванням оптичної густини їх фільтрують у кювету крізь паперовий фільтр.
Розрахунки:
Масову частку вітаміну РР (Х)
у відсотках обчислюють за формулою:
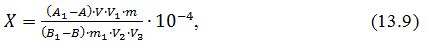
де А1 - значення оптичної
густини випробуваного розчину;
А - значення оптичної густини розчину
для поправки на забарвлені сторонні речовини;
V - загальний об'єм гідролізату,
см3;
V1 - об'єм розчину після очищення цинк сульфатом,
см3;
m- маса вітаміну РР в 5 см3 робочого стандартного розчину,
мкг, m = 10 мкг;
B1- значення оптичної густини робочого стандартного розчину вітаміну РР
(середнє значення з трьох паралельних визначень);
B - значення оптичної густини контрольного
розчину;
m1 - маса наважки, г;
V2- об'єм гідролізату, взятого на очистку,
см3;
V3 - об'єм очищеного гідролізату, взятого для кольорової реакції,
см3.
За остаточний результат випробування
приймають середнє арифметичне значення двох паралельних визначень, розрахованих
до четвертого і округлених до третього десяткового знака, розбіжність між якими
при Р = 0,95 не повинна перевищувати 13% по відношенню до середнього
арифметичного значення.
Мінімальна концентрація вітаміну РР, обумовлена зазначеним методом, становить 0,5 мкг в 1 см3 колориметрованого розчину
13.1.5 Визначення
вітаміну B1 (тіаміну)
Тіамін (вітамін В1) відіграє важливу роль в обміні речовин.
Коферментна похідна тіаміну - тіамінпірофосфат - входить до складу ряду
ферментів, які каталізують окислювальне декарбоксилювання
кетокислот.
13.1.5.1 Визначення
вітаміну B1 (тіаміну) з
використанням хроматографічної колонки
Метод поширюється на харчові консервовані продукти з овочів, фруктів, ягід, овочів з м'ясом, крупами, молоком тощо. Межа виявлення масової частки вітаміну B1 становить 0,008•10-3% за довірчої ймовірності р = 0,95.
Суть методу
Заснована на кислотному і ферментативному гідролізі пов'язаних форм
вітаміну, очищення гідролізату на колонці з катіонітом, окислені тіаміну в
тіохром і вимірюванні інтенсивності флуоресценції за довжин хвиль 320…390 нм
збуджувального і 400…580 нм випромінювального світла.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: терези лабораторні з найбільшою межею зважування 200 г; терези
лабораторні з найбільшою межею зважування 500 г; рН-метр лабораторний з
діапазоном вимірювань величини рН від 4 до 9; термостат, що забезпечує
підтримування температури 37 ° С; флуорометр; електрошафа сушильна
лабораторна; баня водяна; лійки ділильні скляні місткістю 50 або 100
см3; лійки лабораторні скляні діаметром від 56 до 150 мм; колби мірні
лабораторні скляні місткістю 100, 250, 1000 см3; колби лабораторні
скляні по місткістю 250 см3; колонки для хроматографії: складаються
зі з'єднаних між собою скляних трубок різного діаметру; верхня частина колонки
повинна бути довжиною 9…10 см і внутрішнім діаметром 2,5…3,0 см, нижня -
довжиною 14…15 см, внутрішнім діаметром 0,6…0,7 см; колонку необхідно закривати
краном; при відсутності крана, колонка повинна складатися з трьох трубок: дві
верхні трубки повинні мати ті ж параметри, нижня є капілярною трубкою довжиною
2,5…3,0 см, внутрішнім діаметром 0,10…0,15 см; палички скляні; піпетки мірні
лабораторні скляні місткістю 1, 2, 5, 10, 20 см3; прилад для
перегонки на шліфах; пробірки градуйовані скляні на 20, 25 см3;
склянки лабораторні скляні місткістю 50, 200 см3; циліндри мірні
лабораторні скляні місткістю 10, 25, 50, 100, 250 см3; папір
індикаторний універсальний; папір фільтрувальний лабораторний.
Реактиви: вода дистильована; амоній роданистий, розчин з масовою часткою 50%; калій залізосиньородистий, розчин з масовою часткою 1% (зберігають в темній склянці протягом двох днів); кислота хлоридна, розчини з молярною концентрацією с (НСl) = 0,1 моль/дм3 і об'ємною часткою 50%; калій хлористий, розчин з масовою часткою 25% в розчині хлоридної кислоти с (НСl) = 0,1 моль / дм3; кислота оцтова, льодяна і розчин з масовою часткою 3%; натру гідроокис, розчини з масовою часткою 10 і 30%; натр оцтовокислий 3-водний, насичений розчин (готують розчиненням 200 г солі в 300 см3 дистильованої води); катіоніт КРС-1п, КРС-ЗпТ40 або КРС-8п, фракція 0,5…1,0 мм; препарат ферментний амілорізін П10Х; препарат ферментний пектаваморин П10Х; спирт ізобутиловий або н-бутиловий, ізоаміловий (перед використанням вимірюють інтенсивність флуоресценції, за наявності флуоресценції спирт очищають перегонкою); спирт етиловий ректифікований; тіаміну хлорид або тіаміну бромід, розчини з масовими концентраціями 0,1; 0,001; 0,0001 г/дм 3; фермент пепсин.
Підготовка до проведення вимірювань:
- Відбір середньої проби та підготовка її до аналізу виконуюють згідно пункту 7.1.1. Проби, які надходять у лабораторію
повинні бути без пошкоджень і змін якості продукту при транспортуванні та
зберіганні;
- Приготування стандартного розчину тіаміну: тіамін попередньо висушують протягом 2 годин у сушильній шафі при
100 ° С. Для приготування розчину з масовою концентрацією
0,1 г / дм3 розчиняють 0,100 г тіаміну в дистильованої
воді в мірній колбі на 1000 см3, додають 10 крапель концентрованої
хлоридної кислоти, доводять до позначки водою і перемішують. Для приготування
розчину тіаміну з масовою концентрацією 0,001 г / дм3 1,0
см3 розчину з концентрацією 0,1 г/дм3 відбирають у мірну
колбу на 100 см3, доводять до позначки дистильованою водою і
перемішують. Для приготування розчину тіаміну з масовою концентрацією
0,0001 г / дм3 10,0 см3 розчину з
концентрацією 0,001 г / дм3 відбирають у мірну колбу на 100
см3, доводять до позначки дистильованою водою і перемешують. Розчин з
масовою концентрацією 0,1 г / дм3 зберігають у темній
склянці в холодильнику протягом місяця. Розчини з масовими концентраціями 0,01 і
0,0001 г / дм3 готують у день проведення
аналізу.
- Обробка, регенерація катіоніту і заповнення колонок:
невикористаний катионіт попередньо заливають розчином хлоридної кислоти
з об'ємною часткою 50%, витримують протягом доби і промивають декантацією
дистильованою водою до видалення іонів заліза. Відсутність іонів заліза
перевіряють за розчином роданистого амонію. Для цього в склянку місткістю 50
см3 додають 5…10 см3 води після промивання катионіта, 10
крапель розчину хлоридної кислоти з об'ємною часткою 50%, 3 см3
розчину роданистого амонію. Промитий катионіт повинен мати безбарвну реакцію
промивної води. Перед заповненням колонки катіонітом в нижню частину колонки над
краном або над капілярною трубкою поміщають невелику кількість скляної вати. Для
заповнення колонки в неї наливають суміш катіоніту з водою після збовтування.
Висота шару катіоніту в колонці – 9…10 см. Для регенерації катіоніту після
проведення аналізу в колонку наливають розчин гідроксиду натру з масовою часткою
10%, пропускають його до виходу з колонки і потім, закривши кран, витримують
20…30 хв. За відсутності в колонці крана на нижній кінець її має бути надіта
гумова трубка внутрішнім діаметром 0,3…0,4 см і піднята на рівень шару рідини в
колонці. Катіоніт на колонці промивають дистильованою водою до нейтральної
реакції за універсальним індикаторним папером.
- Приготування окиснювальної суміші: готують змішуванням 2,0 см3 розчину залізосиньородистого калію з 10,0 см3 розчину їдкого натру з масовою часткою 30%.
Хід аналізу:
1.
Приготування гідролізату:
- наважку проби масою 50,0 г – для консервованого продукту без додавання
вітамінів, 20,0 г – з додаванням вітамінів переносять в мірну колбу на
250 см3,
- додають 150 см3 розчину хлоридної кислоти с(НСl) = 0,1 моль / дм3;
нагрівають на киплячій водяній бані протягом 40
хв.;
- охолоджують до кімнатної температури і проводять ферментативний гідроліз;
*При аналізі фруктових та ягідних консервів з великим вмістом пектинових речовин (з яблук, агрусу, чорної смородини тощо) використовують ферментний препарат амілорізін П10Х і пектаваморин П10Х. В інших випадках використовують тільки амілорізін П10Х.
- попередньо встановлюють величину рН 4,2-4,5: вміст колби поміщають в
стакан місткістю 200 см3, додають при постійному перемішуванні розчин
натрій ацетату до потрібної величини рН. Вимірювання величини рН виконують
рН-метром, після цього вміст склянки переносять в ту ж саму колбу місткістю 250 см3,
додають 0,10 г амілорізіну П10Х, або по 0,10 г амілорізіну П10Х і пектаваморину
П10Х перемішують, закривають пробкою і витримують в термостаті при
37 ° С протягом 12…16 годин. Гідролізат охолоджують до кімнатної
температури, доводять до позначки дистильованою водою, перемішують і
фільтрують.
- овочеві консерви з м'ясом обробляють ферментами пепсином і амілорізіном
П10Х: в колбу поміщають 0,10 г пепсину і витримують в термостаті при
37 ° С протягом 4 годин. Потім встановлюють величину рН 4,2…4,5,
додають 0,10 г амілорізіну П10Х, перемішують і знову витримують у термостаті за
тієї ж температури протягом 12…16 годин. Після цього гідролізат охолоджують,
доводять до позначки дистильованою водою, перемішують і
фільтрують.
2.Очищення
гідролізату
- перед очищенням гідролізату катіоніт на колонці переводять в Н-форму:
через колонку пропускають 20,0 см3 розчину оцтової кислоти, нагрітої
до температури 60…70 ° С;
- в одну колонку вносять 20,0 см3 гідролізату, в іншу 20,0
см3 стандартного розчину тіаміну з масовою концентрацією 0,0001
г/дм3 (швидкість пропускання через колонку - не менше 15 крапель за 1
хв);
- після проходження розчину катіоніт на колонці промивають дистильованою
водою три рази по 10 см3 і вносять 20,0 см3 розчину калій
хлориду, нагрітого до температури 60…70 ° С;
- збирають 20,0 см3 елюата в градуйовані пробірки або циліндри
місткістю 20 або 25 см3;
3.Окислення тіаміну в тіохром
- у дві ділильні лійки поміщають по 5,0 см3 елюата,
отриманого після очищення гідролізату проби;
- у дві інші – по 5,0 см3 елюата, отриманого після пропускання
через колонку стандартного розчину тіаміну;
- потім у дві лійки (з елюатом проби і стандартного розчину) додають по
1,20 см3 окиснювальної суміші (окислена форма),
- в дві інші (з тими ж розчинами) - по 1,20 см3 розчину їдкого
натру з масовою часткою 30% (неокислена форма);
- вміст лійок перемішують, додають по 10,0 см3 ізобутилового
спирту і струшують протягом 1 хв.;
- для прискорення розшаровування після збовтування додають по 0,50
см3 етилового спирту;
- після розшаровування рідин нижній водний шар видаляють, а верхній
органічний шар зливають у кювету для вимірювання інтенсивності флуоресценції;
- вимірюють спочатку інтенсивність флуоресценції стандартних розчинів (окислену і неокислену форму), потім аналізованих проб.
Розрахунки:
Масову частку вітаміну B1 (Х) у відсотках обчислюють за формулою:

С1 - інтенсивність флуоресценції неокисленої форми аналізованого
розчину;
D -
інтенсивність флуоресценції окисленої форми стандартного
розчину;
D1 - інтенсивність флуоресценції неокисленої форми стандартного
розчину;
а - маса тіаміну в 5 см3
стандартного розчину, взята для окислення, г;
К - коефіцієнт перерахунку тіаміна
броміду на тіаміна хлорид, при використанні для приготування стандартного розчину тіаміна хлориду К = 1; тіаміна броміду К = 1,17;
V - загальний об'єм гідролізату,
см3;
V1 - об'єм елюата, взятий для окислення,
см3;
m - маса наважки продукту, г.
За остаточний результат аналізу приймають середнє арифметичне значення результатів двох паралельних визначень за довірчої ймовірності 0,95, допустиме розходження між якими не повинно перевищувати: 0,004•10- 3% абс. - за масової частки вітаміну B1 не більше 0,050•10-3%; та 0,060•10-3% абс. - за масової частки вітаміну В1 більше 0,050•10-3%.
13.1.5.2 Флюорометричний
метод визначення вітаміну В1 (тіаміну) за А. І. Єрмаковим
Метод поширюється на свіжу рослинну продукцію та продукти її переробки.
Суть методу
Полягає в окиснені тіаміну червоною кров'яною сіллю в лужному
середовищі. При цьому утворюється тіохром, який флюоресцує голубим світлом за
ультрафіолетового освітлення.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали: терези лабораторні з найбільшою межею зважування 200 г; терези лабораторні з найбільшою межею зважування 500 г; ступка з товкачиком, термостат, що забезпечує підтримування температури 37 ° С; флуорометр; трубка для абсорбції (рис. 38); баня водяна; лійки ділильні скляні місткістю 50 або 100 см3; лійки лабораторні скляні діаметром від 56 до 150 мм; колби мірні лабораторні скляні місткістю 100, 250, 1000 см3; колби лабораторні скляні по місткістю 250 см3; палички скляні; піпетки мірні лабораторні скляні місткістю 1, 2, 5, 10, 20 см3; пробірки градуйовані скляні на 20, 25 см3; склянки лабораторні скляні місткістю 50, 200 см3; циліндри мірні лабораторні скляні місткістю 10, 25, 50, 100, 250 см3; папір індикаторний універсальний; папір фільтрувальний лабораторний.
Реактиви: сульфатна кислота (0,1 н
розчин H2SO4), хлоридна кислота (10 % - вий розчин HCl), їдкий натр (15 % - вий розчин NaOH), натрій ацетат (2,5 М розчин), червона кров'яна сіль
(1 % - вий розчин K3[Fe(CN)6], готується в день аналізу); для окиснення витяжки використовують
0,04%-вий розчин K3[Fe(CN)6] у 15 % -вому розчині
NaOH: до 4 см3 вихідного розчину приливають 96 см3
15 % -вого розчину
NaOH, зберігають розчин протягом 4 годин; ацетатний буфер рН 3,8 – 4;
3 % - ва оцтова кислота; 25 % - вий розчин
KCl у 0,1 н розчині HCl; ізобутиловий спирт, який не повинен флюоресцувати (можно очистити
активованим вугіллям: 30 г вугілля на 1 дм3 з подальшою перегонкою в
приладі зі шлифами; при перевірці очищеного спирту на флюориметрі стрілка
гальванометра не повинна відхилятися більше ніж на 5 поділок); адсорбент –
катіонід СДВ – 3 (регенерацію та активування адсорбенту виконують нагріванням
протягом 15 хв. з 10-разовою кількістю 3 %-вої оцтової кислоти за
температури не вище 60…70 °С у зв'язку з термолабільністю); ферментний препарат,
який містить фосфотазу (кисла фосфотаза); стандартний розчин тіаміну: 10 мг
тіаміна броміду або тіаніна хлориду розчиняють у 100 см3 0,01 н
розчину HCl (розчин добре зберігається в холодильнику); із основного розчину у день
визначення готують робочий розчин: 1 см3 стандартного розчину
доводять дистильованою водою до 100 см3, у 1 см3
останнього розчину міститься 1 мкг тіаміна броміда).
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні;
Хід аналізу:
1.Вилучення тіаміну
- наважку подрібненого рослинного матеріалу масою 5…10 г поміщають у
ступку;
- наливають невелику кількість 0,1 н розчину H2SO4 та ретельно розтирають;
- розтерту масу переносять у конічну колбу, так, щоб кінцевий об'єм
дорівнював 50…75 см3;
- вміст колби нагрівають протягом 45 хв. на киплячій водяній
бані;
- після охолодження в колбу додають 2,5 М натрій ацетату до рН 4,5…5 (біля
5 см3) і ферментний препарат фосфатази;
- колби ставлять у термостат при 37 °С на ніч;
- потім вміст колби переносять у мірну колбу на 100 см3,
доводять дистильованою водою до позначки і фільтрують через паперовий
фільтр;
- для адсорбції беруть 10 см3 витяжки.
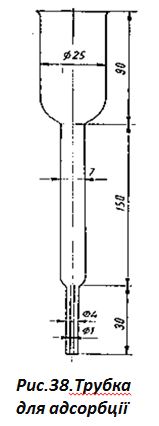
2.Адсорбція тіаміну:
- для проведення адсорбції використовують спеціальні трубки (рис.
38);
- на дно трубки поміщають шматочок вати, насипають 6…8 см
адсорбенту;
- через адсорбент пропускають 10 см3 3 % - вої оцтової кислоти і додають 10 см3
витяжки;
- потім стовпчик адсорбенту тричі промивають 10 см3
дистильованої води (30 см3);
- елюювання тіаміну виконують гарячим 25 % - вим розчином KCl у 0,1 н розчині HCl, невеликими порціями по 5…6 см3;
Визначення вітаміну В1 у ряді обє'ктів допустимо проводити спрощено, без використання адсорбенту. Для цього частину витяжки (20…25 см3), яку отримали після фільтрування початкового екстракту, струшують 2 рази з рівним об'ємом ізобутилового спирту протягом 1…2 хв. Верхній спиртовий шар, який містить небажані домішки, відкидають. Нижній шар, після відшаровування і фільтрування використовують для окиснення. Можливість виключення стадії адсорбції для різних об'єктів визначається експериментальним шляхом.
3.Окиснення дослідних і стандартних розчинів (отримання
тіохрома):
- у ділильні лійки наливають по 5 см3 отриманого елюату (або
витяжки);
- у одну з лійок приливають 3 см3 0,04%-вого розчину K3[Fe(CN)6] у 15 % -вому розчині NaOH, перемішують протягом 30 с та доливають 12 см3 ізобутилового
спирту;
- у другу лійку (контрольну) приливають 3 см3 15 % -вого розчину
NaOH (без солі), перемішують протягом 30 с та доливають 12 см3
ізобутилового спирту;
- ділильні лійки закривають пробками і ретельно струшують протягом 1
хв.;
- після розшаровування нижній шар зливають;
- в ізобутиловий спирт для його просвітлення (сушки) додають 2
см3 96 %-вого етилового спирту, струшують, дають постояти та
прозорий розчин зливають у пробірку для флюоресценції;
- для сушки можна використовувати безводний Na2SO4,
додаючи його в пробірку по 2…2,5 г. Якщо розчин ще мутний, то слід додати натрій сульфату. Для
флюорометрування використовують виключно прозорі розчини.
- одночасно виконують окислення стандартного
розчину;
- для цього у дві ділильні лійки вводять по 1 см3 робочего
розчину і додають 4 см3 води;
- у одну з лійок додають лужний розчин з червоною кров'яною
сіллю;
- в іншу – тільки лужний розчин;
- перемішують протягом 30 с та доливають 12 см3 ізобутилового
спирту;
- ділильні лійки закривають пробками і ретельно струшують протягом 1
хв.;
- після розшаровування нижній шар зливають;
- в ізобутиловий спирт для його просвітлення (сушки) додають 2
см3 96 %-вого етилового спирту, струшують, дають постояти та
прозорий розчин зливають у пробірку для флюоресценції;
4.Визначають інтенсивність флюоресценції всіх
розчинів.
Розрахунки:
Масову частку тіаміну (Х), мкг/г, визначають за формулою:

де
А – показник флюорометра для дослідного розчину;
В -
показник флюорометра для дослідного розчину без
окислювача;
А1
- показник флюорометра для стандартного розчину;
В1
- показник флюорометра для стандартного розчину без
окислювача;
а – 1 мкг тіаміну в 1 см3 стандартного
розчину;
V1
– загальний об'єм витяжки після ферментативного гідролізу, см3,
(звичайно 100 см3);
V2
– об'єм витяжки для визначення (в даному випадку для адсорбції),
см3,
V3
– об'єм елюату, см3, (25 см3);
V4
–
обєм витяжки для окиснення, см3, (5
см3);
H
– маса наважки.
Якщо, визначення проводились без використання адсорбента (очищення
початкової витяжки ізобутиловим спиртом та подальшим окисненням її), то
розрахунок необхідно виконувати за
формулою:

За остаточний результат випробування приймають середнє арифметичне
значення двох паралельних визначень, розрахованих до четвертого і округлених до
третього десяткового знака, розбіжність між якими при Р = 0,95 не
повинна перевищувати 13% по відношенню до середнього арифметичного значення.
13.1.6
Визначення вітаміну В2 (рибофлавіну)
Рибофлавін (вітамін В2) – кристалічна речовина, розчинна у
воді, етиловому, метиловому, аміловому, бутиловому спиртах. Добре розчиняється у
лужних розчинах. Нерозчинний у бензолі, ацетоні, етерах, хлороформі.
У розчинах рибофлавін вважається стійким. Швидкість його руйнування
сильно корелює із величиною рН та інтенсивністю світлового випромінювання. У
лужному середовищі рибофлавін руйнується, у кислому середовищі є стійким при
нагріванні.
У природних об'єктах виявлені 4 форми рибофлавіну: вільний рибофлавін,
флавінмононуклеотид, флавінаденіндинуклеотид та форма, яка міцно пов'язана з
білком і була відкрита К. Л. Поволоцкою.
13.1.6.1 Рибофлавіновий
метод визначення вітаміну В2
Метод призначений для аналізу слабозабарвлених овочевих, фруктових і
ягідних консервованих продуктів. Межа виявлення масової частки вітаміну В2 становить
0,005•10-3%, при довірчій ймовірності 0,95.
Суть методу
Заснована на кислотному і ферментативному гідролізі пов'язаних форм
вітаміну, окисленні пігментів калій перманганатом, відновлені рибофлавіну натрій
гідросульфітом і вимірюванні інтенсивності флуоресценції до і після відновлення
за довжин хвиль 360…480 нм збуджувального і 510…650 нм випромінювального
світла.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали:
терези лабораторні з найбільшою межею зважування 200 г; терези
лабораторні з найбільшою межею зважування 500 г; рН-метр лабораторний з
діапазоном вимірювань величини рН від 4 до 9; термостат, що забезпечує
підтримування температури 37 ° С; флуорометр; електрошафа сушильна
лабораторна; баня водяна; лійки ділильні скляні місткістю 50 або 100
см3; лійки лабораторні скляні діаметром від 56 до 150 мм; колби мірні
лабораторні скляні місткістю 100, 250, 1000 см3; колби лабораторні
скляні по місткістю 250 см3; палички скляні; піпетки мірні
лабораторні скляні місткістю 1, 2, 5, 10, 20 см3; прилад для
перегонки на шліфах; пробірки градуйовані скляні на 20, 25 см3;
склянки лабораторні скляні місткістю 50, 200 см3; циліндри мірні
лабораторні скляні місткістю 10, 25, 50, 100, 250 см3; папір
індикаторний універсальний; папір фільтрувальний лабораторний, ексикатор
вакуумний; колби лабораторні скляні місткістю 50 см3.
Реактиви: вода дистильована; амоній роданистий, розчин з масовою часткою 50%; калій залізосиньородистий, розчин з масовою часткою 1% (зберігають в темній склянці протягом двох днів); кислота хлоридна, розчини з молярною концентрацією с (НСl) = 0,1 моль/дм3 і об'ємною часткою 50%; калій хлористий, розчин з масовою часткою 25% у розчині хлоридної кислоти с (НСl) = 0,1 моль / дм3; кислота оцтова, льодяна і розчин з масовою часткою 3%; натру гідроокис, розчини з масовою часткою 10 і 30%; натрій оцтовокислий 3-водний, насичений розчин (готують розчиненням 200 г солі в 300 см3 дистильованої води); катіоніт КРС-1п, КРС-ЗпТ40 або КРС-8п, фракція 0,5…1,0 мм; препарат ферментний амілорізін П10Х; препарат ферментний пектаваморин П10Х; спирт ізобутиловий або н-бутиловий, ізоаміловий (перед використанням вимірюють інтенсивність флуоресценції, за наявності флуоресценції спирт очищають перегонкою); спирт етиловий ректифікований; тіаміну хлорид або тіаміну бромід, розчини з масовими концентраціями 0,1; 0,001; 0,0001 г/дм3; фермент пепсин, розчин гідроген пероксиду з масовою часткою 3% (термін зберігання сім днів); калій перманганат, розчин з масовою часткою 3%. (термін зберігання сім днів); кислота сульфатна розчин з масовою часткою 93,6…95,6%; натрій гідросульфіт технічний; рибофлавін, розчини з масовими концентраціями 0,02 і 0,001 г/дм3.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні;
Приготування стандартного розчину
рибофлавіну
- попередньо висушують рибофлавін в вакуум-ексикаторі над концентрованої
сульфатної кислотою;
- для приготування стандартного розчину рибофлавіну з масовою
концентрацією 0,02 г/дм3 в мірну колбу місткістю 1000 см3
поміщають 0,020 г рибофлавіну, додають близько 750 см3 дистильованої
води, 1,0 см3 льодяної оцтової кислоти і нагрівають при перемішуванні
до розчинення;
- потім розчин охолоджують до кімнатної температури, доводять до позначки
дистильованою водою і знову перемішують;
- для приготування розчину рибофлавіну з масовою концентрацією 0,001
г/дм3 відбирають 5,0 см3 розчину з масовою концентрацією
0,02 г/дм3 у мірну колбу місткістю 100 см3, доводять до
мітки дистильованою водою і перемішують;
- розчин з концентрацією 0,02 г/дм3 зберігають у холодильнику протягом місяця; розчин з концентрацією 0,001 г/дм3 готують у день проведення аналізу.
Хід аналізу:
1.
Приготування гідролізату
- наважку проби масою 50,0 г – для консервованого продукту без додавання
вітамінів, 20,0 г – з додаванням вітамінів переносять в мірну колбу на
250 см3,
- додають 150 см3 розчину хлоридної кислоти с(НСl) = 0,1 моль / дм3;
- нагрівають на киплячій водяній бані протягом 40 хв.;
- охолоджують до кімнатної температури і проводять ферментативний гідроліз;
*При аналізі фруктових та ягідних консервів з великим вмістом пектинових речовин (з яблук, агрусу, чорної смородини тощо) використовують ферментний препарат амілорізін П10Х і пектаваморин П10Х. В інших випадках використовують тільки амілорізін П10Х.
- попередньо встановлюють величину рН 4,2-4,5: вміст колби поміщають в
стакан місткістю 200 см3, додають при постійному перемішуванні розчин
натрій ацетату до потрібної величини рН. Вимірювання величини рН виконують
рН-метром, після цього вміст склянки переносять в ту ж саму колбу місткістю 250 см3,
додають 0,10 г амілорізіну П10Х, або по 0,10 г амілорізіну П10Х і пектаваморину
П10Х перемішують, закривають пробкою і витримують в термостаті при
37 ° С протягом 12…16 годин.
- гідролізат охолоджують до кімнатної температури, доводять до позначки
дистильованою водою, перемішують і фільтрують.
- овочеві консерви з м'ясом обробляють ферментами пепсином і амілорізіном
П10Х: в колбу поміщають 0,10 г пепсину і витримують в термостаті при
37 ° С протягом 4 годин. Потім встановлюють величину рН 4,2…4,5,
додають 0,10 г амілорізіну П10Х, перемішують і знову витримують у термостаті за
тієї ж температури протягом 12…16 годин. Після цього гідролізат охолоджують,
доводять до позначки дистильованою водою, перемішують і
фільтрують.
Для визначення поправки на вміст вітаміну
В2 у ферментах одночасно проводять контрольний дослід:
- у мірну колбу на 250 см3 поміщають 150 см3 розчину
хлоридної кислоти с(НСl) = 0,1
моль/дм3,
- встановлюють за допомогою розчину натрій ацетату величину рН
4,2…4,5;
- додають 0,10 г амілорізіну П10Х або по 0,10 г амілорізіну П10Х і
пектаваморину П10Х і витримують у термостаті при 37 ° С протягом 12…16
годин;
- при використанні амілорізіну П10Х і пепсину в колбу з розчином хлоридної
кислоти с(НСl) = 0,1 моль/дм3
додають спочатку 0,10 г пепсину і витримують в термостаті 4 години за тієї ж
температури;
- встановлюють величину рН 4,2-4,5,
- додають 0,10 г амілорізіну П10Х і знову витримують 12…16 годин при
37 ° С;
- вміст колби охолоджують, доводять до позначки дистильованою водою,
перемішують і фільтрують.
*Контрольний дослід проводять один раз для кожної партії
ферментів.
- у дві колби на 50 см3 поміщають по 10,0 см3
гідролізату проби, в третю колбу - 10,0 см3 гідролізату контрольного
досліду;
- в одну колбу з аналізованим гидролизатом додають 1,0 см3
стандартного розчину рибофлавіну з концентрацією 0,001 г/дм3,
- в дві інші - по 1,0 см3 дистильованої
води;
- в усі колби додають по 1,0 см3 льодяної оцтової кислоти, 0,50
см3 розчину калій перманганату, перемішують і витримують 2
хв.;
- додають по 0,50 см3 розчину гідроген пероксиду і знову
перемішують;
- через 5 хв. вимірюють інтенсивність флуоресценції спочатку розчину з добавкою рибофлавіну, потім розчину без добавки і контрольного досліду.
**Вимірювання інтенсивності флюоресценції кожного розчину виконують спочатку без додавання натрій гідросульфіту, потім після його додавання. Натрій гідросульфіт додають порціями по 0,02…0,05 г, швидко перемішують і знову виконують вимірювання. Цю операцію повторюють до встановлення найменшої інтенсивності флюоресценції.
Розрахунки:
Масову частку вітаміну В2 (Х) у відсотках обчислюють за формулою:

А1
- інтенсивність флюоресценції розчину аналізованої проби після додавання
натрій гідросульфіту;
С - інтенсивність флюоресценції розчину
контрольного досліду до додавання натрій гідросульфіту;
С1
- інтенсивність флюоресценції розчину контрольного досліду після додавання
натрій гідросульфіту;
D - інтенсивність флюоресценції розчину аналізованої проби з добавкою
рибофлавіну до додавання натрій гідросульфіту;
D1 - інтенсивність флюоресценції розчину аналізованої проби з добавкою
рибофлавіну після додавання натрій гідросульфіту;
b - маса рибофлавіну в стандартному розчині рибофлавіну, додана в
гідролізат аналізованої проби, г;
V - загальний об'єм гідролізату, см3;
V1
- об'єм гідролізату, взятий для окиснення,
см3;
m - наважка продукту, г.
0,95, допустиме розходження між якими не повинно перевищувати: 0,008•10-3% абс. при масовій частці вітаміну В2 не більше 0,050•10-3%; та 0,020•10-3% абс. - при масовій частці вітаміну В2 більше 0,050•10-3%.
13.1.5.2 Флюорометричний
метод визначення вітаміну В1 (тіаміну) за
А. І. Єрмаковим
Суть методу
Полягає в окиснені тіаміну червоною кров'яною сіллю в лужному
середовищі. При цьому утворюється тіохром, який флюоресцує голубим світлом за
ультрафіолетового освітлення.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали: терези лабораторні з найбільшою межею зважування 200 г; терези лабораторні з найбільшою межею зважування 500 г; ступка з товкачиком, термостат, що забезпечує підтримування температури 37 ° С; флуорометр; трубка для абсорбції (рис. 38); баня водяна; лійки ділильні скляні місткістю 50 або 100 см3; лійки лабораторні скляні діаметром від 56 до 150 мм; колби мірні лабораторні скляні місткістю 100, 250, 1000 см3; колби лабораторні скляні по місткістю 250 см3; палички скляні; піпетки мірні лабораторні скляні місткістю 1, 2, 5, 10, 20 см3; пробірки градуйовані скляні на 20, 25 см3; склянки лабораторні скляні місткістю 50, 200 см3; циліндри мірні лабораторні скляні місткістю 10, 25, 50, 100, 250 см3; папір індикаторний універсальний; папір фільтрувальний лабораторний.
Реактиви: сульфатна кислота (0,1 н
розчин H2SO4), хлоридна кислота (10 % - вий розчин HCl), їдкий натр (15 % - вий розчин NaOH), натрій ацетат (2,5 М розчин), червона кров'яна сіль
(1 % - вий розчин K3[Fe(CN)6], готується в день аналізу); для окиснення витяжки використовують
0,04%-вий розчин K3[Fe(CN)6] у 15 % -вому розчині
NaOH: до 4 см3 вихідного розчину приливають 96 см3
15 % -вого розчину
NaOH, зберігають розчин протягом 4 годин; ацетатний буфер рН 3,8 – 4;
3 % - ва оцтова кислота; 25 % - вий розчин
KCl у 0,1 н розчині HCl; ізобутиловий спирт, який не повинен флюоресцувати (можно очистити
активованим вугіллям: 30 г вугілля на 1 дм3 з подальшою перегонкою в
приладі зі шлифами; при перевірці очищеного спирту на флюориметрі стрілка
гальванометра не повинна відхилятися більше ніж на 5 поділок); адсорбент –
катіонід СДВ – 3 (регенерацію та активування адсорбенту виконують нагріванням
протягом 15 хв. з 10-разовою кількістю 3 %-вої оцтової кислоти за
температури не вище 60…70 °С у зв'язку з термолабільністю); ферментний препарат,
який містить фосфотазу (кисла фосфотаза); стандартний розчин тіаміну: 10 мг
тіаміна броміду або тіаніна хлориду розчиняють у 100 см3 0,01 н
розчину HCl (розчин добре зберігається в холодильнику); із основного розчину у день
визначення готують робочий розчин: 1 см3 стандартного розчину
доводять дистильованою водою до 100 см3, у 1 см3
останнього розчину міститься 1 мкг тіаміна броміда).
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні;
Хід аналізу:
1.Вилучення тіаміну
- наважку подрібненого рослинного матеріалу масою 5…10 г поміщають у
ступку;
- наливають невелику кількість 0,1 н розчину H2SO4 та ретельно розтирають;
- розтерту масу переносять у конічну колбу, так, щоб кінцевий об'єм
дорівнював 50…75 см3;
- вміст колби нагрівають протягом 45 хв. на киплячій водяній
бані;
- після охолодження в колбу додають 2,5 М натрій ацетату до рН 4,5…5 (біля
5 см3) і ферментний препарат фосфатази;
- колби ставлять у термостат при 37 °С на ніч;
- потім вміст колби переносять у мірну колбу на 100 см3,
доводять дистильованою водою до позначки і фільтрують через паперовий
фільтр;
- для адсорбції беруть 10 см3 витяжки.
2.Адсорбція тіаміну:
- для проведення адсорбції використовують спеціальні трубки (рис.
38);
- на дно трубки поміщають шматочок вати, насипають 6…8 см
адсорбенту;
- через адсорбент пропускають 10 см3 3 % - вої оцтової кислоти і додають 10 см3
витяжки;
- потім стовпчик адсорбенту тричі промивають 10 см3
дистильованої води (30 см3);
- елюювання тіаміну виконують гарячим 25 % - вим розчином KCl у 0,1 н розчині HCl, невеликими порціями по 5…6 см3;
*Визначення вітаміну В1 у ряді обє'ктів допустимо проводити спрощено, без використання адсорбенту. Для цього частину витяжки (20…25 см3), яку отримали після фільтрування початкового екстракту, струшують 2 рази з рівним об'ємом ізобутилового спирту протягом 1…2 хв. Верхній спиртовий шар, який містить небажані домішки, відкидають. Нижній шар, після відшаровування і фільтрування використовують для окиснення. Можливість виключення стадії адсорбції для різних об'єктів визначається експериментальним шляхом.
3.Окиснення дослідних і стандартних розчинів (отримання
тіохрома):
- у ділильні лійки наливають по 5 см3 отриманого елюату (або
витяжки);
- у одну з лійок приливають 3 см3 0,04%-вого розчину K3[Fe(CN)6] у 15 % -вому розчині NaOH, перемішують протягом 30 с та доливають 12 см3 ізобутилового
спирту;
- у другу лійку (контрольну) приливають 3 см3 15 % -вого розчину
NaOH (без солі), перемішують протягом 30 с та доливають 12 см3
ізобутилового спирту;
- ділильні лійки закривають пробками і ретельно струшують протягом 1
хв.;
- після розшаровування нижній шар зливають;
- в ізобутиловий спирт для його просвітлення (сушки) додають 2
см3 96 %-вого етилового спирту, струшують, дають постояти та
прозорий розчин зливають у пробірку для флюоресценції;
- для сушки можна використовувати безводний Na2SO4,
додаючи його в пробірку по 2…2,5 г. Якщо розчин ще мутний, то слід додати натрій сульфату. Для
флюорометрування використовують виключно прозорі розчини.
- одночасно виконують окислення стандартного
розчину;
- для цього у дві ділильні лійки вводять по 1 см3 робочего
розчину і додають 4 см3 води;
- у одну з лійок додають лужний розчин з червоною кров'яною
сіллю;
- в іншу – тільки лужний розчин;
- перемішують протягом 30 с та доливають 12 см3 ізобутилового
спирту;
- ділильні лійки закривають пробками і ретельно струшують протягом 1
хв.;
- після розшаровування нижній шар зливають;
- в ізобутиловий спирт для його просвітлення (сушки) додають 2
см3 96 %-вого етилового спирту, струшують, дають постояти та
прозорий розчин зливають у пробірку для флюоресценції;
4.Визначають інтенсивність флюоресценції всіх
розчинів.
Розрахунки:
Масову частку тіаміну (Х), мкг/г, визначають за формулою:
В -
показник флюорометра для дослідного розчину без
окислювача;
А1
- показник флюорометра для стандартного розчину;
В1
- показник флюорометра для стандартного розчину без
окислювача;
а – 1 мкг тіаміну в 1 см3 стандартного
розчину;
V1
– загальний об'єм витяжки після ферментативного гідролізу, см3,
(звичайно 100 см3);
V2
– об'єм витяжки для визначення (в даному випадку для адсорбції),
см3,
V3
– об'єм елюату, см3, (25 см3);
V4
–
обєм витяжки для окиснення, см3, (5
см3);
H
– маса наважки.
Якщо, визначення проводились без використання адсорбента (очищення початкової витяжки ізобутиловим спиртом та подальшим окисненням її), то розрахунок необхідно виконувати за формулою:
За остаточний результат випробування приймають середнє арифметичне значення двох паралельних визначень, розрахованих до четвертого і округлених до третього десяткового знака, розбіжність між якими при Р = 0,95 не повинна перевищувати 13% по відношенню до середнього арифметичного значення.
13.1.6 Визначення
вітаміну В2 (рибофлавіну)
Рибофлавін (вітамін В2) – кристалічна речовина, розчинна у
воді, етиловому, метиловому, аміловому, бутиловому спиртах. Добре розчиняється у
лужних розчинах. Нерозчинний у бензолі, ацетоні, етерах, хлороформі.
У розчинах рибофлавін вважається стійким. Швидкість його руйнування сильно корелює із величиною рН та інтенсивністю світлового випромінювання. У лужному середовищі рибофлавін руйнується, у кислому середовищі є стійким при нагріванні.
У природних об'єктах виявлені 4 форми рибофлавіну: вільний рибофлавін, флавінмононуклеотид, флавінаденіндинуклеотид та форма, яка міцно пов'язана з білком і була відкрита К.Л. Поволоцкою.
13.1.6.1 Рибофлавіновий
метод визначення вітаміну В2
Метод призначений для аналізу слабозабарвлених овочевих, фруктових і
ягідних консервованих продуктів. Межа виявлення масової частки вітаміну В2 становить
0,005•10-3%, при довірчій ймовірності 0,95.
Суть методу
Заснована на кислотному і ферментативному гідролізі пов'язаних форм
вітаміну, окисленні пігментів калій перманганатом, відновлені рибофлавіну натрій
гідросульфітом і вимірюванні інтенсивності флуоресценції до і після відновлення
за довжин хвиль 360…480 нм збуджувального і 510…650 нм випромінювального
світла.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали:
терези лабораторні з найбільшою межею зважування 200 г; терези
лабораторні з найбільшою межею зважування 500 г; рН-метр лабораторний з
діапазоном вимірювань величини рН від 4 до 9; термостат, що забезпечує
підтримування температури 37 ° С; флуорометр; електрошафа сушильна
лабораторна; баня водяна; лійки ділильні скляні місткістю 50 або 100
см3; лійки лабораторні скляні діаметром від 56 до 150 мм; колби мірні
лабораторні скляні місткістю 100, 250, 1000 см3; колби лабораторні
скляні по місткістю 250 см3; палички скляні; піпетки мірні
лабораторні скляні місткістю 1, 2, 5, 10, 20 см3; прилад для
перегонки на шліфах; пробірки градуйовані скляні на 20, 25 см3;
склянки лабораторні скляні місткістю 50, 200 см3; циліндри мірні
лабораторні скляні місткістю 10, 25, 50, 100, 250 см3; папір
індикаторний універсальний; папір фільтрувальний лабораторний, ексикатор
вакуумний; колби лабораторні скляні місткістю 50 см3.
Реактиви: вода дистильована; амоній роданистий, розчин з масовою часткою 50%; калій
залізосиньородистий, розчин з масовою часткою 1% (зберігають в темній склянці протягом
двох днів); кислота хлоридна, розчини з молярною концентрацією с (НСl) = 0,1 моль/дм3 і
об'ємною часткою 50%; калій хлористий, розчин з масовою часткою 25% у розчині
хлоридної кислоти с (НСl) = 0,1 моль / дм3;
кислота оцтова, льодяна і розчин з масовою часткою 3%; натру гідроокис, розчини
з масовою часткою 10 і 30%; натрій оцтовокислий 3-водний, насичений розчин
(готують розчиненням 200 г солі в 300 см3 дистильованої води);
катіоніт КРС-1п, КРС-ЗпТ40 або КРС-8п, фракція 0,5…1,0 мм; препарат
ферментний амілорізін П10Х; препарат ферментний пектаваморин П10Х; спирт
ізобутиловий або н-бутиловий,
ізоаміловий (перед використанням вимірюють інтенсивність флуоресценції, за
наявності флуоресценції спирт очищають перегонкою); спирт етиловий
ректифікований; тіаміну хлорид або тіаміну бромід, розчини з масовими концентраціями
0,1; 0,001; 0,0001 г/дм3; фермент пепсин, розчин гідроген пероксиду з масовою часткою 3% (термін зберігання сім
днів); калій перманганат, розчин з масовою часткою 3%. (термін зберігання сім
днів); кислота сульфатна розчин з масовою часткою 93,6…95,6%; натрій
гідросульфіт технічний; рибофлавін, розчини з масовими концентраціями 0,02 і
0,001 г/дм3.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні;
Приготування стандартного розчину рибофлавіну:
- попередньо висушують рибофлавін в вакуум-ексикаторі над концентрованої
сульфатної кислотою;
-для приготування стандартного розчину рибофлавіну з масовою
концентрацією 0,02 г/дм3 в мірну колбу місткістю 1000 см3
поміщають 0,020 г рибофлавіну, додають близько 750 см3 дистильованої
води, 1,0 см3 льодяної оцтової кислоти і нагрівають при перемішуванні
до розчинення;
- потім розчин охолоджують до кімнатної температури, доводять до позначки
дистильованою водою і знову перемішують;
- для приготування розчину рибофлавіну з масовою концентрацією 0,001
г/дм3 відбирають 5,0 см3 розчину з масовою концентрацією
0,02 г/дм3 у мірну колбу місткістю 100 см3, доводять до
мітки дистильованою водою і перемішують;
- розчин з концентрацією 0,02 г/дм3 зберігають у холодильнику протягом місяця; розчин з концентрацією 0,001 г/дм3 готують у день проведення аналізу.
Хід аналізу:
1.Приготування гідролізату
- наважку проби масою 50,0 г – для консервованого продукту без додавання
вітамінів, 20,0 г – з додаванням вітамінів переносять в мірну колбу на
250 см3,
- додають 150 см3 розчину хлоридної кислоти с(НСl) = 0,1 моль / дм3;
- нагрівають на киплячій водяній бані протягом 40 хв.;
- охолоджують до кімнатної температури і проводять ферментативний гідроліз;
*При аналізі фруктових та ягідних консервів з великим вмістом пектинових речовин (з яблук, агрусу, чорної смородини тощо) використовують ферментний препарат амілорізін П10Х і пектаваморин П10Х. В інших випадках використовують тільки амілорізін П10Х.
- попередньо встановлюють величину рН 4,2-4,5: вміст колби поміщають в
стакан місткістю 200 см3, додають при постійному перемішуванні розчин
натрій ацетату до потрібної величини рН. Вимірювання величини рН виконують
рН-метром, після цього вміст склянки переносять в ту ж саму колбу місткістю 250 см3,
додають 0,10 г амілорізіну П10Х, або по 0,10 г амілорізіну П10Х і пектаваморину
П10Х перемішують, закривають пробкою і витримують в термостаті при
37 ° С протягом 12…16 годин.
- гідролізат охолоджують до кімнатної температури, доводять до позначки
дистильованою водою, перемішують і фільтрують.
- овочеві консерви з м'ясом обробляють ферментами пепсином і амілорізіном П10Х: в колбу поміщають 0,10 г пепсину і витримують в термостаті при 37 ° С протягом 4 годин. Потім встановлюють величину рН 4,2…4,5, додають 0,10 г амілорізіну П10Х, перемішують і знову витримують у термостаті за тієї ж температури протягом 12…16 годин. Після цього гідролізат охолоджують, доводять до позначки дистильованою водою, перемішують і фільтрують.
Для визначення поправки на вміст вітаміну В2 у
ферментах одночасно проводять контрольний дослід:
- у мірну колбу на 250 см3 поміщають 150 см3 розчину
хлоридної кислоти с(НСl) = 0,1
моль/дм3,
- встановлюють за допомогою розчину натрій ацетату величину рН
4,2…4,5;
- додають 0,10 г амілорізіну П10Х або по 0,10 г амілорізіну П10Х і
пектаваморину П10Х і витримують у термостаті при 37 ° С протягом 12…16
годин;
- при використанні амілорізіну П10Х і пепсину в колбу з розчином хлоридної
кислоти с(НСl) = 0,1 моль/дм3
додають спочатку 0,10 г пепсину і витримують в термостаті 4 години за тієї ж
температури;
- встановлюють величину рН 4,2-4,5,
- додають 0,10 г амілорізіну П10Х і знову витримують 12…16 годин при
37 ° С;
- вміст колби охолоджують, доводять до позначки дистильованою водою, перемішують і фільтрують.
*Контрольний дослід проводять один раз для кожної партії ферментів
2.Підготовка до вимірювання
- у дві колби на 50 см3 поміщають по 10,0 см3
гідролізату проби, в третю колбу - 10,0 см3 гідролізату контрольного
досліду;
- в одну колбу з аналізованим гидролизатом додають 1,0 см3
стандартного розчину рибофлавіну з концентрацією 0,001 г/дм3,
- в дві інші - по 1,0 см3 дистильованої
води;
- в усі колби додають по 1,0 см3 льодяної оцтової кислоти, 0,50
см3 розчину калій перманганату, перемішують і витримують 2
хв.;
- додають по 0,50 см3 розчину гідроген пероксиду і знову
перемішують;
- через 5 хв. вимірюють інтенсивність флуоресценції спочатку розчину з добавкою рибофлавіну, потім розчину без добавки і контрольного досліду.
**Вимірювання інтенсивності флюоресценції кожного розчину виконують спочатку без додавання натрій гідросульфіту, потім після його додавання. Натрій гідросульфіт додають порціями по 0,02…0,05 г, швидко перемішують і знову виконують вимірювання. Цю операцію повторюють до встановлення найменшої інтенсивності флюоресценції.
Розрахунки:
Масову частку вітаміну В2 (Х) у відсотках обчислюють за формулою:
де А - інтенсивність
флюоресценції розчину аналізованої проби до додавання натрій
гідросульфіту;
А1
- інтенсивність флюоресценції розчину аналізованої проби після додавання
натрій гідросульфіту;
С -
інтенсивність флюоресценції розчину контрольного досліду до додавання натрій
гідросульфіту;
С1
- інтенсивність флюоресценції розчину контрольного досліду після додавання
натрій гідросульфіту;
D - інтенсивність флюоресценції розчину аналізованої проби з добавкою
рибофлавіну до додавання натрій гідросульфіту;
D1 - інтенсивність флюоресценції розчину аналізованої проби з добавкою
рибофлавіну після додавання натрій гідросульфіту;
b - маса рибофлавіну в стандартному розчині рибофлавіну, додана в
гідролізат аналізованої проби, г;
V - загальний об'єм гідролізату, см3;
V1
- об'єм гідролізату, взятий для окиснення,
см3;
m - наважка продукту, г.
За остаточний результат аналізу приймають середнє арифметичне значення
результатів двох паралельних визначень за довірчої ймовірності 0,95, допустиме
розходження між якими не повинно перевищувати: 0,008•10-3% абс. при
масовій частці вітаміну В2 не більше 0,050•10-3%; та
0,020•10-3% абс. - при масовій частці вітаміну В2 більше
0,050•10-3%.
13.1.6.2 Люмінофлавіновий
метод визначення вітаміну В2
Метод призначений для аналізу овочевих консервів з м'ясом і крупами, а також темних консервованих продуктів.
Суть методу
Заснована на кислотному і ферментативному гідролізі пов'язаних форм
вітаміну, окисленні пігментів калій перманганатом, опроміненні світлом для
перетворення рибофлавіну в люміфлавін, екстракції люміфлавіну хлороформом і
вимірюванні інтенсивності флуоресценції за довжин хвиль 360…480 нм
збуджувального і 510…650 нм випромінювального світла.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали: терези лабораторні з найбільшою межею зважування 200 г; терези лабораторні з найбільшою межею зважування 500 г; рН-метр лабораторний з діапазоном вимірювань величини рН від 4 до 9; термостат, що забезпечує підтримування температури 37 ° С; флуорометр; електрошафа сушильна лабораторна; баня водяна; лійки ділильні скляні місткістю 50, 100, 250 см3; лійки лабораторні скляні діаметром від 36 до 150 мм; колби мірні лабораторні скляні місткістю 100, 250, 1000 см3; колби лабораторні скляні по місткістю 250 см3; палички скляні; піпетки мірні лабораторні скляні місткістю 1, 2, 5, 10, 20 см3; прилад для перегонки на шліфах; пробірки градуйовані скляні на 20, 25 см3; склянки лабораторні скляні місткістю 50, 200 см3; циліндри мірні лабораторні скляні місткістю 10, 25, 50, 100, 250 см3; папір індикаторний універсальний; папір фільтрувальний лабораторний, ексикатор вакуумний; колби лабораторні скляні місткістю 50 см3, вентилятор побутовий; лампа розжарювання електрична загального призначення, потужністю 100, 150, 200 Вт; бюретки місткістю 50, 100 см3; колби лабораторні скляні плоскодонні або конічні з взаємозамінним конусом на 100 см3.
Реактиви: вода дистильована; амоній роданистий, розчин з масовою часткою 50%; калій
залізосиньородистий, розчин з масовою часткою 1% (зберігають в темній склянці протягом
двох днів); кислота хлоридна, розчини з молярною концентрацією с (НСl) = 0,1 моль/дм3 і
об'ємною часткою 50%; калій хлористий, розчин з масовою часткою 25% в розчині
хлоридної кислоти с (НСl) = 0,1 моль / дм3;
кислота оцтова, льодяна і розчин з масовою часткою 3%; натрій гідроксид, розчини
з масовою часткою 10 і 30%; натрій оцтовокислий 3-водний, насичений розчин
(готують розчиненням 200 г солі в 300 см3 дистильованої води);
катіоніт КРС-1п, КРС-ЗпТ40 або КРС-8п, фракція 0,5…1,0 мм; препарат
ферментний амілорізін П10Х; препарат ферментний пектаваморин П10Х; спирт
ізобутиловий або н-бутиловий,
ізоаміловий (перед використанням вимірюють інтенсивність флуоресценції, за
наявності флуоресценції спирт очищають перегонкою); спирт етиловий
ректифікований; тіаміну хлорид або тіаміну бромід, розчини з масовими концентраціями
0,1; 0,001; 0,0001 г/дм3; фермент пепсин, розчин перекису водню з масовою часткою 3% (термін зберігання сім днів);
калій перманганат, розчин з масовою часткою 3%. (термін зберігання сім днів);
кислота сульфатна розчин з масовою часткою 93,6…95,6%; натрій гідросульфіт
технічний; рибофлавін, розчини з масовими концентраціями 0,02 і 0,001
г/дм3, кислота сульфатна, розчин з масовою часткою 30%; натрій
гідроксид, розчин з молярною концентрацією с(NaOH) = 7 моль/дм3; натрій
сульфатний, безводний; хлороформ технічний (перед використанням вимірюють
інтенсивність флюоресценції, за наявності очищають
перегонкою).
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін якості продукту при транспортуванні та зберіганні;
Приготування стандартного розчину рибофлавіну
- попередньо висушують рибофлавін в вакуум-ексикаторі над концентрованою
сульфатною кислотою;
- для приготування стандартного розчину рибофлавіну з масовою
концентрацією 0,02 г/дм3 в мірну колбу місткістю 1000 см3
поміщають 0,020 г рибофлавіну, додають близько 750 см3 дистильованої
води, 1,0 см3 льодяної оцтової кислоти і нагрівають при перемішуванні
до розчинення;
- потім розчин охолоджують до кімнатної температури, доводять до позначки
дистильованою водою і знову перемішують;
- для приготування розчину рибофлавіну з масовою концентрацією 0,001
г/дм3 відбирають 5,0 см3 розчину з масовою концентрацією
0,02 г/дм3 у мірну колбу місткістю 100 см3, доводять до
мітки дистильованою водою і перемішують;
- розчин з концентрацією 0,02 г/дм3 зберігають у холодильнику протягом місяця; розчин з концентрацією 0,001 г/дм3 готують у день проведення аналізу.
Хід аналізу:
1.Приготування гідролізату
- наважку проби масою 50,0 г – для консервованого продукту без додавання
вітамінів, 20,0 г – з додаванням вітамінів переносять у мірну колбу на
250 см3,
- додають 150 см3 розчину хлоридної кислоти с(НСl) = 0,1 моль / дм3;
- нагрівають на киплячій водяній бані протягом 40 хв.;
- охолоджують до кімнатної температури і проводять ферментативний
гідроліз;
*При аналізі фруктових та ягідних консервів з великим вмістом пектинових речовин (з яблук, аґрусу, чорної смородини тощо) використовують ферментний препарат амілорізін П10Х і пектаваморин П10Х. В інших випадках використовують тільки амілорізін П10Х.
- попередньо встановлюють величину рН 4,2-4,5: вміст колби поміщають в
стакан місткістю 200 см3, додають при постійному перемішуванні розчин
натрій ацетату до потрібної величини рН. Вимірювання величини рН виконують
рН-метром, після цього вміст склянки переносять в ту ж саму колбу місткістю 250 см3,
додають 0,10 г амілорізіну П10Х, або по 0,10 г амілорізіну П10Х і пектаваморину
П10Х перемішують, закривають пробкою і витримують в термостаті при
37 ° С протягом 12…16 годин.
- гідролізат охолоджують до кімнатної температури, доводять до позначки
дистильованою водою, перемішують і фільтрують.
- овочеві консерви з м'ясом обробляють ферментами пепсином і амілорізіном П10Х: в колбу поміщають 0,10 г пепсину і витримують в термостаті при 37 ° С протягом 4 годин. Потім встановлюють величину рН 4,2…4,5, додають 0,10 г амілорізіну П10Х, перемішують і знову витримують у термостаті за тієї ж температури протягом 12…16 годин. Після цього гідролізат охолоджують, доводять до позначки дистильованою водою, перемішують і фільтрують.
Для визначення поправки на вміст вітаміну В2 у
ферментах одночасно проводять контрольний дослід:
- у мірну колбу на 250 см3 поміщають 150 см3 розчину
хлоридної кислоти с(НСl) = 0,1
моль/дм3,
- встановлюють за допомогою розчину натрій ацетату величину рН
4,2…4,5;
- додають 0,10 г амілорізіну П10Х або по 0,10 г амілорізіну П10Х і
пектаваморину П10Х і витримують у термостаті при 37 ° С протягом 12…16
годин;
- при використанні амілорізіну П10Х і пепсину в колбу з розчином хлоридної
кислоти с(НСl) = 0,1 моль/дм3
додають спочатку 0,10 г пепсину і витримують в термостаті 4 години за тієї ж
температури;
- встановлюють величину рН 4,2-4,5,
- додають 0,10 г амілорізіну П10Х і знову витримують 12…16 годин при
37 ° С;
- вміст колби охолоджують, доводять до позначки дистильованою водою,
перемішують і фільтрують.
2.Очищення гідролізату
- у колбу місткістю 250 см3 поміщають 100 см3
аналізованого гідролізату,
- в іншу - 100 см3 гідролізату контрольного
досліду;
- в обидві колби додають по 2,0 см3 розчину сульфатної
кислоти;
- піпеткою з поділками по краплях при постійному перемішуванні додають
розчин калій перманганату до слабко-рожевого забарвлення, яке не зникає протягом
1 хв.;
- надлишок рожевого забарвлення усувають додаванням по краплях розчину
гідроген пероксиду;
- відзначають витрачені об'єми розчинів калій перманганату і гідроген
пероксиду;
- отриманий розчин переносять у ділильну лійку місткістю 250
см3, додають 30-50 см3 хлороформу і струшують 0,5
хв.;
- хлороформовий шар видаляють, а водний екстракт використовують для
аналізу.
3.Опромінення екстракту і отримання
люміфлавіну
- у дві колби на 100 см3 додають по 20,0 см3
аналізованого екстракту, в третю - екстракт контрольного
досліду;
- у колбу з аналізованим екстрактом додають по 2,0 см3
стандартного розчину рибофлавіну масовою концентрацією 0,001
г/дм3;
- потім в усі колби додають по 4,0 см3 розчину натрій
гідроксиду з молярною концентрацією с(NaOH) = 7 моль / дм3;
- вміст перемішують, колби закривають пришліфованими пробками і
опромінюють світлом електролампи потужністю 100…200 Вт з відстані 30
± 1 см протягом 40 хв.;
- температура розчину повинна бути
15…25 ° С;
- для охолодження використовують настільний
вентилятор;
- після опромінення в колби додають по 4,0 см3 льодяної оцтової
кислоти, перемішують,
- додають по 20,0 см3 хлороформу і знову перемішують протягом
0,5 хв.;
- вміст колб переносять в ділильні лійки місткістю 50
см3;
- після розшаровування хлороформовий розчин фільтрують у кювети для
вимірювання інтенсивності флуоресценції;
- для зневоднення хлороформного розчину на лійку з фільтром поміщають
близько 2,5 г безводного натрій сульфату;
- вимірюють інтенсивність флюоресценції: спочатку розчину з добавкою
рибофлавіну, потім розчину без добавки і контрольного
досліду.
Розрахунки:
Масову частку вітаміну В2 (Х) у відсотках обчислюють за формулою:
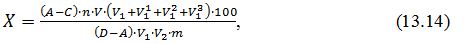
де А - інтенсивність
флюоресценції хлороформового екстракту аналізованої проби;
С - інтенсивність флюоресценції
хлороформового екстракту контрольного досліду;
D - інтенсивність флюоресценції
хлороформового екстракту аналізованої проби з додаванням
рибофлавіну;
n - маса рибофлавіну в стандартному розчині, додана в екстракт
аналізованої проби, г;
V - загальний об'єм гідролізату, см3;
V1 - об'єм гідролізату, взятий для окиснення,
см3;
V11 - об'єм розчину сульфатної кислоти з масовою часткою 30%, доданий
в гідролізат перед окисленням, см3;
V12 - об'єм розчину калію перманганату, витрачений на окиснення
гідролізату, см3;
V13 - об'єм розчину гідроген пероксиду, який витрачено на усунення
надлишку калій перманганату, см3;
V2 - об'єм екстракту, взятий для опромінення, см3;
m - маса наважки продукту, г.
За остаточний результат аналізу приймають середнє арифметичне значення
результатів двох паралельних визначень з довірчою ймовірністю 0,95, допустиме
розходження між якими не повинно перевищувати 0,010•10-3% абс. - за
масової частки вітаміну В2 не більше 0,050•10-3%; та
0,070•10-3% абс. - за масової частки вітаміну В2 більш
0,050•10-3%.
13.1.6.3 Флюорометричний
метод визначення вітаміну В2 (рибофлавіну) за
А. І. Єрмаковим
Суть методу
Заснована на здатності рибофлавіну флюоресціювати в ультрафіолетовому
світлі. Величину флюоресценції вимірюють на флюорометрі.
При визначенні сумарного рибофлавіну враховуються вільний,
мононуклеотидний і динуклеотидний (неміцно і міцно пов'язаний з білком)
рибофлавін.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: флюорометр; терези; мірні циліндри; фарфорова ступка; лійки; складчастий
фільтр; темна склянка з притертою пробкою; мірні колби місткістю 250 см3
; мірні колби місткістю 100 см3; піпетки; термостат; водяна
баня; зворотній холодильник; конічні колби зі шліфом; прилад для струшування;
пробіркі або кювети, гальванометр.
Реактиви: стандартний розчин рибофлавіну; 3 % - вий розчин
Н2О2; 4 % - й розчин КМnО4 (розчин готують через кожні 2 тижні); 2,5 М розчин натрій
ацетату; фосфатний буфер з рН 7,8-8; ферментні препарати: трипсин, панкреатин
або ферментний препарат з міцелію гриба Penicillium (міцелій звільняють від
зайвої вологи під пресом і сушать при 45 ° С; всі зазначені препарати
можна використовувати як протеолітичні, в якості же фосфатазних слід
використовувати тільки останній), ферментний препарат вносят з розрахунку 30
мг / г сухої речовини наважки; 2,5 М оцтовокислий натр; 0,1 н розчин
H2S04.
-
основний розчин станум хлориду:основний розчин готують розчиненням 10 г SnCl2 в 25 см3
концентрованої НС1 і зберігають в темній склянці з притертою пробкою за
кімнатної температури;
- робочий розчин станум
хлориду: готують кожен раз в день визначення, розбавляючи
дистильованою водою 0,2 см3 вихідного розчину до 100
см3);
- розчин натрій
гідросульфіту: 0,25 г Na2S2О4
•2H2O розчиняють в 10 см3 2% - го розчину
NaHCО3; розчин готують в день використання);
- 5 М розчин калій фосфату: 69,6 г К2НРО4 розчиняють в 100 см3
води, якщо при зберіганні розчину випадають кристали, то його перед
використанням нагрівають;
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконуюють згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні;
Приготування стандартного розчину
рибофлавіну
- наважку рибофлавіну масою 10 мг розчиняють в 0,05 н H2S04 у мірній колбі на 250 см3 (в 1 см3
такого розчину міститься 40 мкг рибофлавіну. Розчин стійкий протягом місяця при
зберіганні в темряві і на холоді.);
- робочий розчин готують в день визначення: в мірну колбу на 100
см3 вносять 1 см3 вихідного стандартного розчину і
доводять дистильованою водою до позначки.
При визначенні рибофлавіну з використанням трихлороцтової
кислоти (ТХО) робочий стандартний
розчин для відповідності
дослідному
повинен бути приготований наступним
чином:
- у мірну колбу місткістю 100 см3 вносять 37,5 см3
20 % -вого розчину ТХО,
- додають 24 см3 4 М розчину К2НР04, 1
см3 вихідного стандартного розчину рибофлавіну;
- об`єм доводять дистильованою водою до позначки (в 1 см3 такого робочого розчину міститься 0,4 мкг рибофлавіну).
Хід аналізу:
1.
Приготування гідролізату
- наважку досліджуваного матеріалу (3…5 г) ретельно розтирають у
порцеліновій ступці з невеликою кількістю фосфатного буферу (рН 7,8 - 8);
- розтерту масу переносять у конічну колбу за допомогою буферу з таким
розрахунком, щоб загальне розведення було 1:20;
- суміш витримують на киплячій водяній бані протягом 45 хв.;
- потім охолоджують до 30 ° С, перевіряють рівень рН (він
повинен бути 7,8- 8);
- додають ферментний препарат (трипсин або пакреатин);
- суміш
витримують у
термостаті при 37 оС
протягом 12…16
годин (за таких умов відщеплюється
міцно зв'язана
з білком форма рибофлавіну).
- витяжку
переносять у мірну колбу на 100 см3 і доводять дистильованою водою до
позначки та фільтрують через складчастий фільтр;
- беруть 5 см3 фільтрату в конічну колбу зі шліфом і додають 5
см3 20 % - вого
розчину ТХО;
- колби закривають зворотнім холодильником і витримують на киплячій
водяний бані протягом 10 хв. (обробка ТХО звільняє рибофлавін від його
нуклеотидних форм).
- витяжку
охолоджують і додають 1/5
об'єму
4 М розчину К2НР04 з метою доведення рН до 6.
2.Окиснення
калій перманганатом
- до
витяжки по краплях додають 4 % - вий
розчин КМnО4
до тих пір, поки червонувате забарвлення не перестане зникати;
- через
10 хв.
для руйнування надлишку калій перманганату
по краплях додають, повільно збовтуючи, 3 % -вий
розчин Н2О2;
- до знебарвленого розчину додають 0,2 см3 робочого розчину
SnCl2 і 0,1 см3 розчину натрій
гідросульфіту;
- суміш інтенсивно струшують 20 хв. на приладі для струшування, при цьому
колби пробками не закривають;
- після проведення окиснення і відновлення об'єм витяжки вимірюють і
доводять до 15 см3 дистильованою водою;
- якщо витяжка каламутна, її фільтрують.
3.Вимірювання інтенсивності флюоресценціі
- дослідний і стандартний робочий розчин наливають в пробіркі або кювети з
нефлюоресцируючого скла і вимірюють інтенсивність їх флюоресценції на
флюорометрі;
- після цього в кожну пробірку додають приблизно по 0,1 г
NaHCO3 та 0,1 г
Na2S2О4 • 2Н2О (гідросульфіту
натру) для гасіння флюоресценції рибофлавіну і знову вимірюють флюоресценцію
(домішок).
Іноді виникає необхідність додавати натрій гідросульфіт 2…3 рази (до тих
пір, поки показання гальванометра не стануть постійними).
Розрахунки:
Вміст рибофлавіну Х, мкг/г, визначають за формулою:

де
А - показання флюорометра для дослідного розчину;
В - показання флюорометра для дослідного розчину після гасіння
флюоресценції;
0,4 - вміст рибофлавіну в 1 см3 робочого стандартного
розчину, мкг;
A1 - показання флюорометру для стандартного
розчину;
B1 - показання флюорометру для стандартного розчину після
гасіння флюоресценції (зазвичай близько 0);
V - загальний об'єм витяжки (100 см3);
V1 - об'єм витяжки, взятої для аналізу при кислотному
гідролізі (5 см3); V2 - об'єм екстракту перед вимірюванням
флюоресценції (15 см3);
m – маса наважки рослинного матеріалу, г.
За остаточний результат аналізу приймають середнє арифметичне значення
результатів двох паралельних визначень з довірчою ймовірністю 0,95, допустиме
розходження між якими не повинно перевищувати 0,010•10-3%.
13.2 Визначення вмісту
біологічних пігментів
Біологічні пігменти або біохроми – це забарвлені речовини, що входять до складу тканин рослинних організмів.
Колір пігментів визначається наявністю в них молекул хромофорних групп, які
вибірково поглинають світло в певній частині видимого спектру сонячного світла.
Пігментна система живих рослинного організму – це ланка, яка пов'язує
світлові умови навколишнього середовища і обмін речовин
організму.
Для вищих рослин найбільш важливими вважаються два зелені пігменти:
- хлорофіл “a”, який має зеленувато-синій колір і є основним пігментом
хлоропластів;
- хлорофіл “b”, зеленувато-жовтого кольору, який входить до складу хлоропластів і
переносить засвоєну ним енергію на хлорофіл “a”.
Найчастіше
співвідношення молекул хлорофілу “а”
і “b” в хлоропластах становить 3:1.
Крім того у рослинах наявні каротиноїди. Вони мають різний колір: жовтий, червоний, помаранчевий. Найбільш поширеним з групи каротиноїдів є каротин β.
13.2.1 Спектрофотометричний метод визначення вмісту хлорофілів та каротиноїдів
Вміст
хлорофілів а,
b
та каротиноїдів можна визначити в загальному екстракті пігментів без
попереднього їх розділення.
Для
екстрагування хлорофілів з рослинних тканин використовують молярні розчинники
або суміш полярних (спирт, ацетон) та неполярних (петролейний етер, гексан,
бензин) розчинників. Оскільки пігменти швидко вицвітають на світлі, їх
екстракцію проводять в затемненому приміщенні попередньо охолодженими
розчинниками. Щоб запобігти ізомеризації пігментів, екстракцію слід проводити
якомога швидше. Пігменти екстрагують послідовно кількома порціями розчинника,
фільтруючи кожний раз екстракт крізь скляний фільтр (фільтри Шота
№№ 3, 4). При
розтиранні листків додають невелику кількість МgСО3
або СаСО3 для нейтралізації
кислот клітинного соку з метою запобігання феофітинізації
пігментів.
Визначення
точної концентрації хлорофілів а,
b у
розчині без їх розділення складне, оскільки спектри обох хлорофілів перекривають
один одного,
й неможливо знайти дві довжини хвилі, в яких поглинання обумовлювалося б
повністю лише одним пігментом. Однак існуючі відміни в спектрах поглинання
хлорофілів а
та b
дозволяють вибрати точки, де поглинання одного пігменту суттєво перевищує
поглинання іншого. Цю обставину й використовують при проведенні кількісною
визначення обох хлорофілів без їх розподілу. Однак з двох пігментів точніше
визначається хлорофіл а. В залежності від природи розчинника, що використовують для екстрагування
пігментів, їх концентрації (у мг/л)
розраховують за відповідними рівняннями, наведеними нижче. Останні одержані на
основі експериментально отриманих питомих коефіцієнтів поглинання (С
-
концентрація, D - оптична густина).

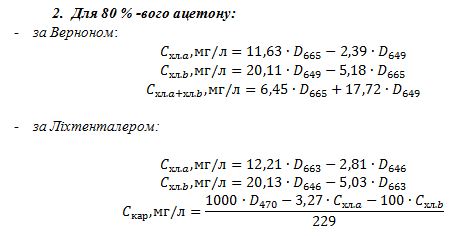

Суть методу
Полягає у вимірюванні оптичної густини витяжки (екстракту) пігментів на
спектрофотометрі за довжин хвиль, що відповідають максимумам поглинання
хлорофілів a і b, та каротиноїдів, з подальшим розрахунком концентрації пігментів за
наведеними вище рівняннями.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали:
кварцовий пісок, порцелянові ступки з товкачиками, скальпель, ножиці,
пінцет, скляні палички, колба Бунзена зі скляним фільтром № 3, мірні колби
об'ємом 25 см3, мірний циліндр об'ємом 25 см3, скляні
лійки, мірні конічні пробірки, піпетки об'ємом 2 та 5 см3, фольга,
терези торсійні, насос Камовського або інший.
Реактиви: етиловий спирт (96 % - вий розчин), ацетон
(80 % - вий розчин) або інший розчинник, MgCO3 або СаСО3, вода дистильована.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконують згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні;
Хід аналізу:
- наважку рослинного матеріалу (100…200 мг) подрібнюють ножицями,
поміщають у порцелянову ступку;
- до матеріалу додають на кінчику скальпеля невелику кількість
MgCO3 (або СаСО3);
- доливають 4…5 см3 ацетону і ретельно
розтирають;
- отриманий екстракт зливають по паличці на скляний фільтр, вставлений у
колбу Бунзена;
- за допомогою насоса рідину відсмоктують;
-після цього в ступку приливають ще невелику кількість ацетону,
розтирають, виливають на фільтр та відсмоктують;
- промивання повторюють декілька разів, доки відфільтрований розчин, не
буде повністю безбарвним;
- екстракт переливають у мірну колбу об'ємом 25
см3,
- колбу Бунзена споліскують декілька разів невеликими порціями ацетону,
який додають до екстракту;
- потім об'єм екстракту доводять чистим ацетоном до позначки (25
см3);
- отриманий ацетоновий екстракт містить суму зелених та жовтих
пігментів.
- визначають оптичну густину (D) екстрактів за довжин хвиль, які відповідають максимумам поглинання
хлорофілів a і b, для 80 % - вого ацетону - 663 та 646 нм, а для
визначення суми каротиноїдів D екстракту вимірюють за λ= 470 нм.
Розрахунки:
Концентрацію хлорофілів a і b та каротиноїдів розраховують за відповідними формулами, наведеними
вище. Потім обов'язково розраховують вміст пігментів (Х) у рослинному матеріалі, мг/г сухої чи
сирої маси, за формулою:

де
С - концентрація пігментів, мг/дм3,
V - об'єм екстракту, см3 (25
см3),
m - наважка рослинного матеріалу, г (0,1…0,2
г).
За остаточний результат аналізу приймають середнє арифметичне значення
результатів двох паралельних визначень з довірчою ймовірністю 0,95, допустиме
розходження між якими не повинно перевищувати 0,010•10- 3%.
13.2.2 Визначення вмісту каротиноїдів
Метод поширюється на функціональні харчові продукти рослинного і
тваринного походження і дає можливість визначення масової концентрації або
масової частки каротиноїдів, що включають загальні каротиноїди і їх окремі
фракції (каротини, ксантофіли, кріптоксантин).
Діапазон вимірювання масової концентрації каротиноїдів становить від 1
до 300 мг / дм3 (кг) (загальні каротиноїди в розрахунку на β-каротин), масової частки - від 1 до
300 мг / дм3 (кг) (загальні каротиноїди в розрахунку на β-каротин), масової частки окремих
фракцій каротиноїдів - від 3% до 80% від загальної кількості
каротиноїдів.
При проведенні досліджень беруть до утерези, що каротиноїди чутливі до
світлових і теплових впливів. Визначення проводять в місці, захищеному від
прямого сонячного або ультрафіолетового випромінювання.
Суть методу
Полягає у екстракції каротиноїдів з проби або осаду, попередньо
отриманого шляхом обробки проби розчинами Карреза I і Карреза II, подальшому
очищенні виділеного препарату петролейним етером і спектрофотометричним
визначенням масової концентрації або масової частки каротиноїдів.
Частки окремих каротиноїдів (від загального вмісту каротиноїдів)
визначають спектрофотометричним вимірюванням у фракціях, одержаних у ході
хроматографічного розділення екстракту.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і матеріали:
випарювач ротаційний вакуумний, спектрофотометр, що дозволяє проводити
вимірювання при 450 нм, терези лабораторні, дозатори автоматичні калібровані або
піпетки скляні калібровані, колонка хроматографічна скляна довжиною 25…30 см і з
розміром внутрішнього діаметра 2 см, гомогенізатор лабораторний механічний для
твердих, рідких і пастоподібних і рідких продуктів, центрифуга, пробірки для центрифугування місткістю 60-100
см3, лійка ділильна місткістю 250 см3, колба мірна
місткістю 100 см3, колба круглодонна на 250 см3, колба
конічна з притертою пробкою на 250 см3,
скловата.
Реактиви: вода дистильована, ацетон, толуол, етер петролейний, алюміній оксид для
колонкової хроматографії, 1-го ступеня активності, ч.д.а., калій
залізистосинеродистий 3-водний, цинк сульфат 7-водний, натрій сульфат безводний,
магній вуглекислий основний водний.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконують згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні;
Приготування розчину Карреза I: 15 г калію залізистосинеродистого 3-водного вносять в мірну колбу
місткістю 100 см3, додають 50 см3 дистильованої води,
ретельно перемішують до повного розчинення. Об’єм розчину в колбі доводять
дистильованою водою до позначки; термін зберігання розчину - не більше 1
міс.
Приготування розчину Карреза II: 30 г цинк сульфату 7-водного вносять в мірну колбу на 100
см3, додають 50 см3 дистильованої води, ретельно
перемішують до повного розчинення. Об’єм розчину в колбі доводять дистильованою
водою до позначки; термін зберігання розчину - не більше 1
міс.
Приготування елюента А: у скляній ємності змішують петролейний етер і толуол в об'ємному
співвідношенні 4: 1; термін зберігання розчину - не більше 1
міс.
Приготування елюента Б: у скляній ємності змішують петролейний етер і толуол в об'ємному
співвідношенні 2: 1; термін зберігання розчину - не більше 1
міс.
Часткова деактивація алюміній оксиду: у конічну колбу з притертою пробкою вносять 100 г активованого алюміній
окису. Додають 12 см3 дистильованої води, ретельно перемішують шляхом
енергійного струшування до отримання тонко дисперсної суспензії. Колбу з
деактивованим алюміній оксидом щільно закривають притертою пробкою. Термін
зберігання розчину - не більше 1 міс.
Підготовка хроматографічної колонки: 28…30 г деактивованого алюміній оксиду суспендують в 30 см3 петролейного етеру. На дно хроматографічної колонки поміщають шматочок скловати. Колонку частково заповнюють петролейним етером. В колонку повільно вносять суспензію алюміній оксиду. Після короткої витримки в колонці повинен сформуватися шар алюміній оксиду висотою близько 8 см. Надмірний об’єм петролейного етеру випускають з колонки. Кінцевий рівень петролейного етеру повинен бути трохи вище поверхні шару алюміній оксиду. Для захисту розділяючого шару в колонці від утворення в ньому повітряних бульбашок, температури етеру і алюміній оксиду не повинні істотно відрізнятися.
Хід аналізу:
1.
Екстрагування каротиноїдів з проб рослинного
походження:
Даний спосіб екстрагування застосовують для виділення каротиноїдів з
твердих, пюре- і пастоподібних продуктів рослинного походження без жирових і
білкових інгредієнтів.
- 2…5 г продукту поміщають в стакан гомогенізатора, додають 100
см3 ацетону і 0,1 г магній карбонату, гомогенізують проби протягом 5
хв.;
- пробу витримують для формування осаду,
- надосадову рідину - екстракт - декантують в ділильну лійку місткістю 250
см3;
- осад послідовно промивають трьома порціями ацетону по 25
см3;
- надосадові рідини кожного разу видаляють з осаду і змішують у ділильній
лійці з екстрактом, який отримано в результаті гомогенізації аналізованої
проби;
- загальний екстракт у ділильній лійці використовують для очищення
органічної фази за допомогою петролейного етеру.
2.Екстрагування каротиноїдів з проб тваринного походження і рідких
функціональних харчових продуктів
Даний спосіб екстрагування застосовують для виділення каротиноїдів з
твердих, пюре- і пастоподібних функціональних харчових продуктів тваринного
походження, що містять жирові і білкові інгредієнти, а також з рідких
функціональних харчових продуктів (наприклад, безалкогольних напоїв, соковмісних
напоїв, соків, морсів, нектарів, коктейлів, тощо).
- за необхідності пробу подрібнюють за допомогою відповідного пристрою і
ретельно перемішують;
- 5…50 г продукту вносять в пробірку для центрифугування місткістю від 60
до 100 см3, додають дистильовану воду до об'єму 50 см3,
ретельно перемішують скляною паличкою;
- до проби додають по 1 см3 розчину Карреза I і Карреза II,
перемішують скляною паличкою, витримують 2 хв. за кімнатної температури;
- вміст пробірки центрифугують протягом 5 хв.
При аналізі рідких продуктів
- в пробірку для центрифугування вносять від 5 до 50 см3 проби
продукту;
- за необхідності об'єм проби продукту в пробірці доводять дистильованою
водою до 50 см3;
- до рідкої проби додають по 1 см3 розчину Карреза I і Карреза
II, перемішують скляною паличкою, витримують 2 хв. за кімнатної
температури;
- вміст пробірки центрифугують протягом 5 хв.
- після центрифугування безбарвну надосадову рідину видаляють з
пробірки;
- до осаду в пробірці додають 40 см3 ацетону, ретельно
перемішують вміст скляною паличкою протягом 3 хв.;
- проводять центрифугування екстракту протягом 5 хв, потім відділяють
надосадову рідину і кількісно переносять її в ділильну лійку місткістю 250
см3;
- осад в пробірці послідовно промивають трьома порціями ацетону по 25
см3, кожен раз проводячи центрифугування для розділення осаду і
екстракту;
- фракції екстракту збирають в ділильній лійці;
- загальний екстракт у ділильній лійці використовують для очищення
органічної фази за допомогою петролейного етеру.
3.Очищення екстракту
- у ділильну лійку додають 50 см3 петролейного етеру, вміст
лійки перемішують і витримують протягом короткого часу для формування верхнього
шару органічної фази;
- у лійку вносять 50 см3 дистильованої води для промивання
органічної фази;
- вміст лійки обережно перемішують легким струшуванням; після витримки
водну фазу видаляють з лійки;
- органічну (петролейну) фазу кількісно переносять із ділильної лійки в
пробірку для центрифугування;
- в пробірку додають 2 г натрій сульфату, вміст ретельно перемішують
скляною паличкою, потім проводять центрифугування для відділення петролейної
фази від осаду;
- після центрифугування петролейну фазу переносять у мірну колбу місткістю
100 см3;
- до осаду в пробірці для центрифугування додають 30 см3
петролейного етеру, перемішують скляною паличкою, повторно
центрифугують;
- відокремлюють петролейну фракцію і додають її до першої порції в мірній
колбі;
- об'єм екстракту в мірній колбі доводять до позначки петролейним етером. Отриманий екстракт використовують для спектрофотометричного визначення загальних каротиноїдів.
*При спектрофотометричному вимірюванні при 450 нм оптична густина отриманого екстракту не повинна перевищувати 0,500 од. Необхідний об’єм аналізованої проби необхідно підбирати емпіричним шляхом.
Так, наприклад, при дослідженні апельсинового соку використовують 25 см3 проби, апельсинового нектару - 50 см3, соку маракуї- від 5 до 20 см3, нектару з маракуї - 25 см3, мандаринового соку - 20 см3.
4.Спектрофотометричне визначення загальних каротиноїдів
Оптичну густину екстракту, вимірюють при 450 нм у спектрофотометрі в
кюветі з оптичного скла і довжиною оптичного шляху 1 см3. В якості
розчину порівняння використовують петролейний етер. Розрахунок масової
концентрації або масової частки загальних каротиноїдів проводять за
формулою (13.16) або (13.17).
5.Хроматографічне розділення фракцій окремих
каротиноїдів
5.1.Концентрування екстракту
- екстракт, переносять у круглодонну колбу ротаційного вакуумного
випарювача місткістю 250 см3;
- розчинник видаляють з екстракту за температури
40 ° С;
- залишок екстрактивних речовин у колбі розчиняють у декількох кубічних
сантиметрах петролейного етеру, потім кількісно переносять до хроматографічної
колонки;
- для виключення втрат круглодонну колбу промивають 10 см3
петролейного етеру, потім також додають отриманий залишковий екстракт до
хроматографічної колонки;
5.2.Фракціювання каротиноїдів
Для хроматографічного розділення екстракту і отримання фракцій окремих
каротиноїдів використовують такі елюенти:
- для каротинової фракції (фракція I) - петролейний
етер;
- фракції кріптоксантинових етерів (фракція II) - елюент
А;
- фракції ксантофілових етерів (фракція III) - елюент
Б;
- фракції залишкових каротиноїдів (фракція IV) - ацетон.
Швидкість елюювання фракції з колонки становить 2 краплі за секунду.
Кількість каротиноїдів у фракціях залежить від їх вмісту в продукті. При
аналізі продуктів рослинного походження (фруктів, напоїв на основі
фруктової сировини) для отримання I-шої та II-гої фракцій потрібно
близько 50 см3 елюента. При високому вмісті каротиноїдів у фракції II
потрібно не більше 100 см3 елюента А. Фракцію III отримують при витраті
від 80 до 100 см3 елюента Б. Для зменшення загального об’єму елюату перші
безбарвні порції розчину відкидають. Для забезпечення повного вилучення
кожної фракції елюювання проводять до тих пір, поки елюат не стане
безбарвним. Залишкові каротиноїди (фракція IV) вилучають із колонки
ацетоном.
6.Спектрофотометричне визначення каротиноїдів у
фракціях
Оптичну густину фракцій окремих каротиноїдів, вимірюють при 450 нм
у спектрофотометрі в кюветі з оптичного скла і довжиною оптичного шляху 1 см. В
якості розчину порівняння використовують петролейний етер.
7.Визначення добавок барвників
Загальна масова частка всіх каротиноїдів в отриманих фракціях повинна
становити не менше 90% від загального вмісту каротиноїдів. Якщо масова частка
каротиноїдів у фракціях менше вказаного значення, то аналізована проба може
містити додані барвники, наприклад біксин. На присутність біксину вказує фракція
червоно-жовтого кольору, яка залишається в хроматографічній колонці після
елюювання останньої фракції каротиноїдів - фракції IV. Біксин може бути
елюйований з колонки сумішшю, яка складається з чотирьох об'ємних частин ацетону
і однієї об'ємної частини розчину амоніаку.
8.Визначення доданих каротиноїдів
Даний метод може бути використаний для визначення вмісту каротиноїдів,
наприклад провітаміну А, доданих у функціональний харчовий продукт. Однак, при
визначенні, в якості контролю використовують паралельну пробу аналізованого
харчового продукту, яка не містить штучний провітамін А.
Розрахунки:
Масову концентрацію каротиноїдів С, мг / дм3 і окремих каротиноїдів у фракціях I-IV розраховують за формулою:

де 4,000 - коефіцієнт перерахунку оптичної
густини;
А - виміряне значення
оптичної густини;
F - фактор розбавлення (відношення об'єму екстракту в петролейному
етері, см3, або об’ємів різних фракцій до об’ємів продукту, взятого
для аналізу, см3.
Масову частку каротиноїдів, Х, мг / кг, у функціональних харчових продуктах розраховують за формулою:

де V- об’єм екстракту в петролейному етері,
см3;
m- маса наважки продукту, г.
За остаточний результат аналізу приймають середнє арифметичне значення
результатів двох паралельних визначень з довірчою ймовірністю 0,95, допустиме
розходження між якими не повинно перевищувати 0,010•10- 3%.
13.2.3 Спектрофотометричний метод визначення каротину і
лікопіну
Лікопін – пігмент з групи каратиноїдів, потужний антиоксидант. Лікопін надає
томатам червоний колір. Найкраще засвоюється лікопін, який потрапляє в організм
із термічно оброблених продуктів: томатний сік, соуси та кетчуп є кориснішими,
ніж салат із помідорів. Високим вмістом лікопіну характеризуються рожевий
грейпфрут, кавун, гострий червоний перець, папая.
Суть методу
Полягає у вимірюванні оптичної густини розчину каротиноїдів при двох
варіантах довжин хвиль: 451 нм (максимум поглинання каротину) і 503 нм (максимум
поглинання лікопіну) і подальшому розрахунку вмісту каротину і лікопіну за
формулами 13.19 та 13.20.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: гомогенізатор; терези лабораторні; ступка порцелянова з товкачиком;
мірні колби на 50, 100 см3; лійка Бюхнера; мірний циліндр; ділильна
лійка місткістю 100…250 см3; дозатори автоматичні калібровані або піпетки скляні калібровані;
спектрофотометр, колби круглодонні на 100, 250 см3.
Реактиви: ацетон; натрій карбонат Na2CO3; петролейний етер
або гексан; натрій сульфатний; 85 %о- вий розчин етанолу;
20 % - вий спиртовий розчин лугу.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконують згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні;
Хід аналізу:
- середню пробу плодів подрібнюють на гомогенізаторі;
- наважку 1…5г розтирають з ацетоном (1:10) у ступці в присутності
невеликої кількості Na2CO3;
- розтерту наважку переносять у колбу на 50…100 см3 і залишають
в темряві на 30 хв. (при серійних визначеннях можна залишити в холодильнику на
ніч);
- окрашенний розчин і осад переносять на лійку Бюхнера і багаторазово
промивають його ацетоном до повного знебарвлення;
- каротин з ацетонової витяжки переводять у петролейний етер або
гексан;
- останній відмивають водою від ацетона в ділильній лійці (зазвичай на
20…30 см3 гексану досить 200…300 см3
води),
- сушать, пропускаючи через колонку з натрій
сульфатом;
- вимірюють об'єм і приступають до
спектрофотометрування.
- якщо плоди містять хлорофіл, то для видалення його гексановий екстракт
промивають 85о% - вим розчином етанолу, омилюють
20% - вим спиртовим розчином лугу, потім ще раз промивають
85о% - вим етанолом;
- гексановий екстракт ретельно відмивають водою від етанолу і лугу,
висушують безводним сірчанокислим натром і визначають вміст
пігментів.
Розрахунки
Кількість каротину Х1 (мг / 100 г)
розраховують для каротину за формулою:

де D - величина оптичної
густини за даної довжини хвилі;
d - товщина поглинаючого шару, см;
с - концентрація вихідної речовини, г / 1000 см3
гексанового екстракту.
За остаточний результат аналізу приймають середнє арифметичне значення результатів двох паралельних визначень за довірчої ймовірності 0,95, допустиме розходження між якими не повинно перевищувати 0,010•10- 3%.
13.3 Визначення вмісту фенольних
речовин
13.3.1 Метод визначення
вмісту поліфенолів за реактивом Фоліна-Деніса
Даний метод визначення вмісту поліфенолів (суми фенольних сполук) може
бути використаний для аналізу їстівної частини фруктів, овочів, а також
продуктів їх переробки.
Суть методу
Грунтується на реакції комплексоутворення поліфенолів з реактивом Фоліна
– Деніса і утворенні забарвлених речовин із наступним визначенням оптичної
густини.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: фотоколориметр або спектрофотометр, терези лабораторні, шафа сушильна з
діапазоном нагрівання від 40 до 200 °С, пробірки скляні, ексикатор, холодильник
скляний зворотний, піпетки поградуйовані 0,5; 1,0; 2,0; 5,0; 20 см3;
годинник пісочний, лійки скляні, палички скляні, стакани, колби конічні, колби
мірні, циліндри, папір фільтрувальний.
Реактиви: вода дистильована, натрій вольфрамат, х.ч.; кислота
фосфорномолібденова, х.ч.; натрій карбонат, насичений розчин; спирт етиловий
ректифікований з масовою часткою 75 % і 95 %; кислота ортофосфорна, ч.д.а.;
рутин, стандартний розчин; кверцетин, стандартний розчин.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконують згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні;
Готування стандартного розчину
- 50 мг кристалічного рутину, попередньо висушеного у сушільній шафі
протягом 180 хв. за температури від 130 °С до 135 °С, розчиняють у 95% - вому етиловому
спирті в мірній колбі на 100 см3., нагріваючи на водяній
бані;
- охолоджують і доводять розчинником до позначки;
- 1 см3 цього розчину містить 0,5 мг
рутину.
Готування реактиву Фоліна – Деніса
- в конічну колбу поміщають 10 г солі вольфрамової кислоти, 2 г
фосфорномолібденової кислоти та 5 см3 ортофосфорної
кислоти;
- ретельно змішують з 75 см3 дистильованої
води;
- реактив поміщають на киплячу водяну баню і витримують зі зворотним
холодильником 120 хв.;
- реактив охолоджують і кількісно переносять у мірну колбу на 100
см3;
- доводять об'єм до позначки дистильованою водою;
- реактив зберігають у темній склянці, у темному місці за температури не вищої 25 °С. Строк придатності реактиву не більше 3 місяців.
Будування калібрувального графіка за
рутином
Готують серію стандартних забарвлених
розчинів:
- в ряд мірних колб додають 1,0; 2,0; 3,0; 4,0; 5,0 см3
стандартного розчину і доводять об'єм до 50 см3 дистильованою
водою;
- у ці колби додають по 5 см3 реактиву
Фоліна-Деніса;
- через 3 хв. додають 10 см3 насиченого розчину натрій карбонату;
- доводять об'єм дистильованою водою до 100 см3, ретельно
струшують і витримують протягом 60 хв.
Отримані розчини містять відповідно 0,050; 0,100; 0,150; 0,200; 0,250 мг рутину в 10 см3.
Приготування контрольного розчину
- у мірну колбу місткістю 100 см3 поміщають ту саму кількість
реактивів, але замість стандартних розчинів додають дистильовану воду, доводять
об'єм до 100 см3.
Через 60 хв. визначають оптичну густину на спектрофотометрі або
фотоелектроколориметрі з довжиною хвилі 670 нм або від 720 нм до 730 нм проти
контрольного розчину. Використовують кювети з відстанню між робочими гранями 10
мм.
Будують калібрувальний графік залежності оптичної густини розчину від
масової концентрації рутину (мг/см3)D=f(c). Новий калібрувальний графік будують при приготуванні нового реактиву
Фоліна – Деніса.
Визначення коефіцієнта екстинції
- у зв'язку з тим, що екстинція змінюється, її слід визначати раз у три
місяці або у разі зміни реактивів;
- для цього вимірюють оптичну густину трьох стандартних забарвлених
розчинів, приготовлених як описано вище, наприклад, з масовою концентрацією
0,10; 0,15; 0,20 мг/см3;
- коефіцієнт екстинції Е визначають за формулою:
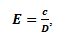
D
– оптична густина розчину відповідної масової
концентрації.
Середній коефіцієнт екстинції Еср визначають з трьох величин, підрахованих до четвертого десяткового знака, розбіжність між якими не повинна перевищувати 10% середнього значення трьох визначень.
Хід аналізу:
1.Отримання екстракту
- з середньої проби продукту беруть наважку від 2
г до 5
г (залежно від передбачуваного вмісту поліфенолів) з похибкою ±
0,01
г;
- поміщають її у конічну колбу місткістю 50
см3;
- \ заливають 75
% етиловим спиртом, нагрітим до температури 60
°С;
- колбу з пробою для іннактивації ферментів, витримують протягом 15
хв. зі зворотним холодильником на водяній бані за температури
80 °С;
- після охолодження екстракт кількісно переносять у мірну колбу на
50
см3 доводять 75
% етиловим спиртом до позначки;
- отриманий екстракт ретельно перемішують і фільтрують.
2.Визначення вмісту поліфенолів
2.1 Приготування дослідного розчину (D):
- 1 см3 екстракту переносять у пробірку;
- додають 7,5 см3 дистильованої води, 0,5 см3
реактиву Фоліна-Деніса;
- через 3 хв. додають 1 см3 насиченого розчину натрій карбонату;
- ретельно струшують і витримують протягом 60 хв. у темному місці.
2.2 Приготування розчину для контролювання кольорових речовин
(DK):
- у пробірку поміщають 1 см3 екстракту;
- додають 8 см3 дистильованої води, 1 см3 насиченого
розчину натрій карбонату;
- ретельно струшують та витримують протягом 60 хв. у темному місці.
2.3 Приготування контрольного розчину (К):
- у пробірку поміщають 8,5
см3 дистильованої води,
- додають 0,5 см3 реактиву Фоліна-Деніса і 1
см3 насиченого розчину натрій
карбонату;
- ретельно струшують та витримують протягом 60 хв. у темному
місці.
- через 60 хв. виконують фотоелектроколориметричне або спектрофотометричне вимірювання усіх трьох розчинів наступним чином: оптичну густину дослідного розчину (D) вимірюють проти контрольного розчину (К), а оптичну густину розчину для контролю кольорових речовин (DK) проти дистильваної води, тобто:
Вимірювання здійснюють за λ = 670 нм, або на фотоелектроколориметрі за
λ = 720…730 нм. Використовують кювети з відстанню між робочими гранями
10 мм.
Розрахунки:
Оптичну густину проби визначають, за формулою:

У разі наявності калібрувального графіка масову частку поліфенолів Х, в
мг на 100 г продукту,
обчислюють за формулою:

де
с
– масова концентрація рутину (або іншої поліфенольної сполуки), яку визначають
за калібрувальним графіком, мг/см3;
V — об'єм екстракту,
см3;
V1 —
об'єм екстракту для аналізу,
см3;
m —
маса наважки продукту, г
100
— коефіцієнт визначення
вмісту поліфенолів на 100 г продукту;
а
— коефіцієнт ступеня розведення екстракту проби.
У разі відсутності калібрувального графіка масову частку поліфенолів Х, в
мг на 100 г продукту, обчислюють за формулою:

де
Епр
— середній коефіцієнт екстинкції;
Dпр—
оптична густина екстракту проби.
За остаточний результат випробувань беруть середнє арифметичне значення
двох паралельних вимірювань, підрахованих до четвертого десяткового знака.
13.3.2 Метод визначення
вмісту поліфенолів в ультрафіолетовій частині спектра
Суть методу
Грунтується на вимірюванні оптичної густини екстрактів
поліфенольновмісної сировини та продуктів її переробки в ультрафіолетовій
частині спектра з довжиною хвилі 280 нм.
Засоби вимірювання, допоміжне обладнання, посуд, реактиви і
матеріали: спектрофотометр, що забезпечує вимірювання з довжиною хвилі від 270 до
280 нм; терези лабораторні, шафа сушильна з діапазоном нагрівання від 40 до 200
°С, водяна баня, пробірки скляні, ексикатор, піпетки поградуйовані 0,5; 1,0;
2,0; 5,0; 20 см3; годинник пісочний, лійки скляні, палички скляні,
стакани, колби конічні, колби мірні, циліндри, папір фільтрувальний.
Реактиви: вода дистильована; спирт етиловий ректифікований з масовою часткою 20%,
75 % і 95 %; рутин, стандартний розчин; кверцетин, стандартний розчин.
Підготовка до проведення вимірювань
Відбір середньої проби та підготовка її до аналізу виконують згідно
пункту 7.1.1.
Проби, які надходять у лабораторію повинні бути без пошкоджень і змін
якості продукту при транспортуванні та зберіганні;
Готування стандартного розчину
- 15 мг кристалічного кверцетину, попередньо висушеного у сушільній шафі
протягом 180 хв. за температури 130 °С, розчиняють у 95% - вому
етиловому спирті в мірній колбі на 50 см3;
- доводять розчинником до позначки;
- 1 см3 цього розчину містить 0,3 мг
кверцетину;
Робочий розчин готують безпосередньо перед будуванням калібрувального графіка
наступним чином:
- 2 см3 стандартного розчину доводять у мірній колбі на 25
см3 до позначки розчином з масовою часткою етилового спирту
50%;
- 1 см3 робочого розчину містить 0,024 мг кверцетину.
Будування калібрувального графіка за
рутином
Готують серію стандартних забарвлених розчинів:
- в 6 пробірок вводять відповідно 0,5; 1,0; 1,5; 2,0; 2,5; 3,0 см3 робочого
розчину і доводять об'єм до 6 см3 20 % -вим етиловим спиртом;
Отримані розчини містять відповідно 0,012; 0,024; 0,036; 0,048; 0,060; 0,072 мг
кверцетину в 1 см3.
- ретельно струшують;
- вимірюють оптичну густину на спектрофотометрі в присутності
20 % - вого спирту з довжиною хвилі 280 нм. Використовують кювети
з відстанню між робочими гранями 10 мм.
- будують калібрувальний графік залежності оптичної густини розчину від
масової концентрації кверцетину (мг/см3)D=f(c);
- будування калібрувального графіка повторюють не менше одного разу у
шість місяців.
Визначення коефіцієнта екстинції
- у зв'язку з тим, що екстинція змінюється, її слід визначати раз у три
місяці або у разі зміни реактивів;
- для цього вимірюють оптичну густину трьох робочих розчинів розчинів,
приготовлених як описано вище, наприклад, з масовою концентрацією 0,036; 0,048;
0,060 мг/см3;
- коефіцієнт екстинції Е визначають за формулою:
де с – масова концентрація
кверцетину (або іншої поліфенольної сполуки),
мг/см3;
D
– оптична густина розчину відповідної масової
концентрації.
Середній коефіцієнт екстинції Еср визначають з трьох величин,
підрахованих до четвертого десяткового знака, розбіжність між якими не повинна
перевищувати 10% середнього значення трьох визначень.
Хід аналізу:
1.Отримання екстракту
- з середньої проби продукту беруть наважку від 2
г до 5
г (залежно від передбачуваного вмісту поліфенолів) з похибкою ±
0,01
г;
- поміщають її у конічну колбу місткістю 50
см3;
- заливають 75
% етиловим спиртом, нагрітим до температури 60
°С;
- колбу з пробою для іннактивації ферментів, витримують
протягом 15
хв. зі зворотним холодильником на водяній бані за температури
80 °С;
- після охолодження екстракт кількісно переносять у мірну колбу на
50
см3 доводять 75
% етиловим спиртом до позначки;
- отриманий екстракт ретельно перемішують і фільтрують.
2.Визначення вмісту поліфенолів
- у пробірки переносять від 0,5 до 3 см3 екстракту і доводять
обєм до 6 см3 дистильованою водою;
- визначають оптичну густину розчину проти дистильованої води за
λ = 280 нм; виористовують кювети з відстанню між робочими гранями
10 мм.
Розрахунки:
У разі наявності калібрувального графіка масову частку поліфенолів Х, в
мг на 100 г продукту,
обчислюють за формулою: